
Genetic Markers of Genome Rearrangements in Helicobacter pylori


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Materials and Identification of Rearrangements
2.2. Identification of Sequence Repeats
2.3. Genomic Islands
3. Results and Discussion
3.1. Genome Rearrangements
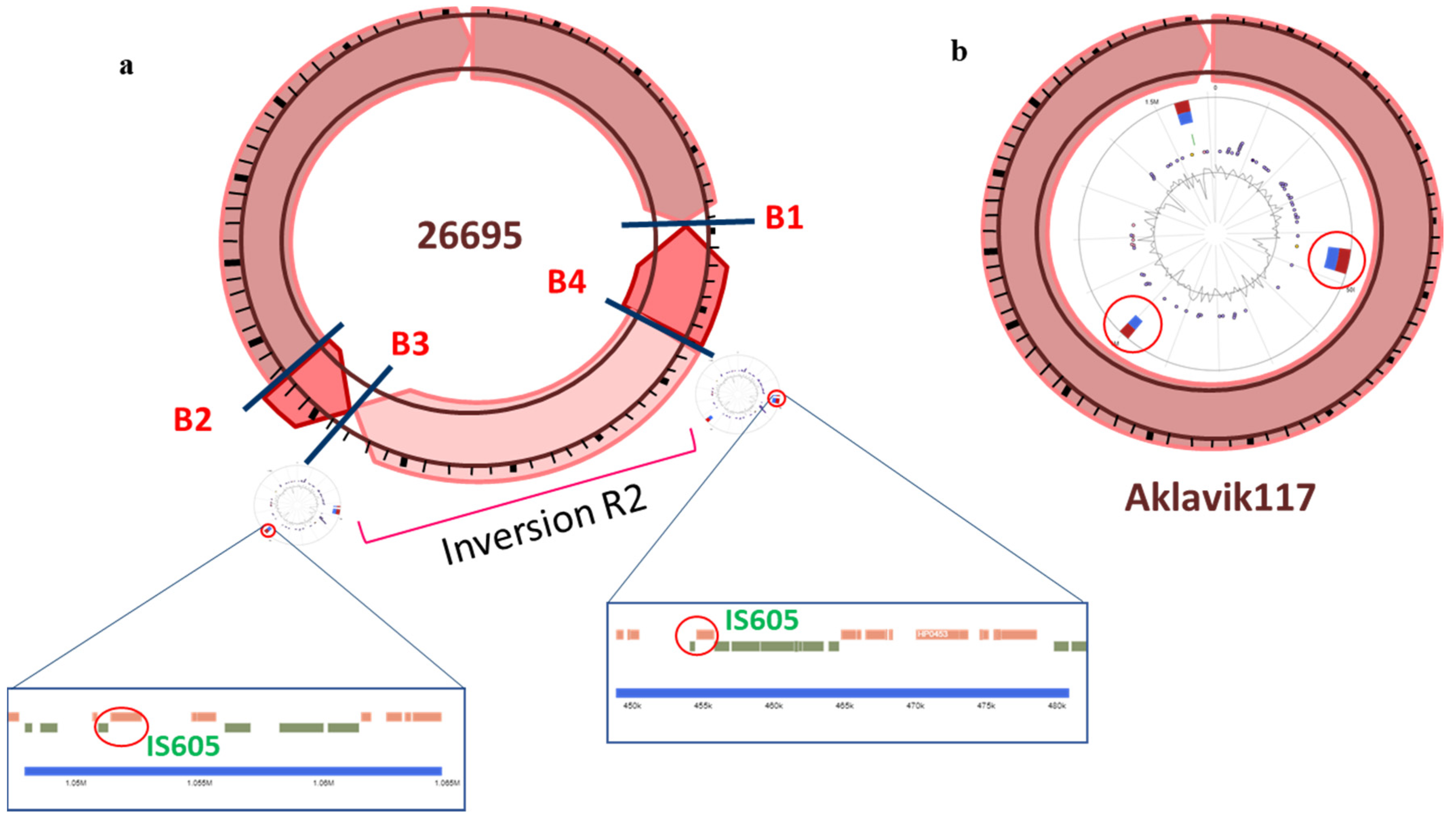
3.2. Inversion Breakpoints
3.3. Repeat Sequences and Their Associated Inversions
3.4. Presence of Genomic Islands around Inversion Breakpoints
3.5. Distribution of Insertion Sequences and Their Association with Inversions
3.6. Other Molecular Elements Related to Inversions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalali, B.; Mejías-Luque, R.; Javaheri, A.; Gerhard, M. H. pylori virulence factors: Influence on immune system and pathology. Mediat. Inflamm. 2014. [Google Scholar] [CrossRef] [Green Version]
- Suerbaum, S.; Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 2002, 347, 1175–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Agency for Research on Cancer (IARC). Schistosomes, Liver Flukes and Helicobacter Pylori, Monograph on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 1994; Volume 61. [Google Scholar]
- Kusters, J.G.; Van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moodley, Y.; Linz, B.; Yamaoka, Y.; Windsor, H.M.; Breurec, S.; Wu, J.Y.; Maady, A.; Bernhöft, S.; Thiberge, J.M.; Phuanukoonnon, S.; et al. The peopling of the Pacific from a bacterial perspective. Science 2009, 323, 527–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linz, B.; Balloux, F.; Moodley, Y.; Manica, A.; Liu, H.; Roumagnac, P.; Falush, D.; Stamer, C.; Prugnolle, F.; van der Merwe, S.W.; et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature 2007, 445, 915–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suerbaum, S.; Josenhans, C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 2007, 5, 441–452. [Google Scholar] [CrossRef]
- Humbert, O.; Dorer, M.S.; Salama, N.R. Characterization of Helicobacter pylori factors that control transformation frequency and integration length during inter-strain DNA recombination. Mol. Microbiol. 2011, 79, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Periwal, V.; Scaria, V. Insights into structural variations and genome rearrangements in prokaryotic genomes. Bioinformatics 2015, 31, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahillon, J.; Chandler, M. Insertion sequences. Microbiol. Mol. Biol. Rev. 1998, 62, 725–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treangen, T.J.; Abraham, A.L.; Touchon, M.; Rocha, E.P. Genesis, effects and fates of repeats in prokaryotic genomes. Fems Microbiol. Rev. 2009, 33, 539–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, E.P.; Danchin, A.; Viari, A. Functional and evolutionary roles of long repeats in prokaryotes. Res. Microbiol. 1999, 150, 725–733. [Google Scholar] [CrossRef]
- Achaz, G.; Coissac, E.; Netter, P.; Rocha, E.P. Associations between inverted repeats and the structural evolution of bacterial genomes. Genetics 2003, 164, 1279–1289. [Google Scholar] [PubMed]
- Zivanovic, Y.; Lopez, P.; Philippe, H.; Forterre, P. Pyrococcus genome comparison evidences chromosome shuffling-driven evolution. Nucleic Acids Res. 2002, 30, 1902–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooka, T.; Ogura, Y.; Asadulghani, M.; Ohnishi, M.; Nakayama, K.; Terajima, J.; Watanabe, H.; Hayashi, T. Inference of the impact of insertion sequence (IS) elements on bacterial genome diversification through analysis of small-size structural polymorphisms in Escherichia coli O157 genomes. Genome Res. 2009, 19, 1809–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajaraman, A.; Tannier, E.; Chauve, C. FPSAC: Fast phylogenetic scaffolding of ancient contigs. Bioinformatics 2013, 29, 2987–2994. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Li, S.; Guo, F.; Ning, K.; Wang, L. Core-genome scaffold comparison reveals the prevalence that inversion events are associated with pairs of inverted repeats. BMC Genom. 2017, 18, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersulyte, D.; Akopyants, N.S.; Clifton, S.W.; Roe, B.A.; Berg, D.E. Novel sequence organization and insertion specificity of IS605 and IS606: Chimaeric transposable elements of Helicobacter pylori. Gene 1998, 223, 175–186. [Google Scholar] [CrossRef]
- Kersulyte, D.; Mukhopadhyay, A.K.; Shirai, M.; Nakazawa, T.; Berg, D.E. Functional organization and insertion specificity of IS607, a chimeric element of Helicobacter Pylori. J. Bacteriol. 2000, 182, 5300–5308. [Google Scholar] [CrossRef] [Green Version]
- Kersulyte, D.; Velapatiño, B.; Dailide, G.; Mukhopadhyay, A.K.; Ito, Y.; Cahuayme, L.; Parkinson, A.J.; Gilman, R.H.; Berg, D.E. Transposable element ISHp608 of Helicobacter pylori: Nonrandom geographic distribution, functional organization, and insertion specificity. J. Bacteriol. 2002, 184, 992–1002. [Google Scholar] [CrossRef] [Green Version]
- Kersulyte, D.; Kalia, A.; Zhang, M.; Lee, H.K.; Subramaniam, D.; Kiuduliene, L.; Chalkauskas, H.; Berg, D.E. Sequence organization and insertion specificity of the novel chimeric ISHp609 transposable element of Helicobacter Pylori. J. Bacteriol. 2004, 186, 7521–7528. [Google Scholar] [CrossRef] [Green Version]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noureen, M.; Tada, I.; Kawashima, T.; Arita, M. Rearrangement analysis of multiple bacterial genomes. BMC Bioinform. 2019, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Ugene Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Rocha, E.P.; Danchin, A.; Viari, A. Analysis of long repeats in bacterial genomes reveals alternative evolutionary mechanisms in Bacillus subtilis and other competent prokaryotes. Mol. Biol. Evol. 1999, 16, 1219–1230. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Huang, H.V. Homologous recombination in Escherichia coli: Dependence on substrate length and homology. Genetics 1986, 112, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Achaz, G.; Rocha, E.P.; Netter, P.; Coissac, E. Origin and fate of repeats in bacteria. Nucleic Acids Res. 2002, 30, 2987–2994. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inversion Type | Total Inversions | IR Associated Inversions | DR Associated Inversions |
|---|---|---|---|
| World-wide | 16 | 5 | 2 |
| Region-specific | 7 | 4 | 1 |
| Strain-specific | 18 | 5 | 3 |
| Region | Average Size of Longest Inverted Repeat | Average Size of Longest Direct Repeat |
|---|---|---|
| East Asia | 2181 | 3425 |
| South America | 1631 | 4495 |
| North America | 2145 | 3745 |
| Europe | 2268 | 2756 |
| Africa | 4587 | 5057 |
| India | 1772 | 4084 |
| Australia | 2315 | 3033 |
| Unknown | 1357 | 3154 |
| Strain | Accession | No. of GIs | GIs Associated with Breakpoints |
|---|---|---|---|
| UM037 | NC_021217.3 | 3 | 1 |
| F32 | NC_017366.1 | 1 | 1 |
| 26695-1CL | NZ_AP013356.1 | 2 | 2 |
| 26695-1CH | NZ_AP013355.1 | 3 | 2 |
| 26695-1 | NZ_AP013354.1 | 3 | 2 |
| Aklavik117 | NC_019560.1 | 4 | 2 |
| 26695-1 | NZ_CP010435.1 | 3 | 2 |
| 26695-1MET | NZ_CP010436.1 | 3 | 2 |
| ELS37 | NC_017063.1 | 2 | 1 |
| Rif2 | NC_018938.1 | 3 | 2 |
| Rif1 | NC_018937.1 | 3 | 2 |
| 26695 | NC_018939.1 | 3 | 2 |
| 26695 | NC_000915.1 | 2 | 2 |
| Gambia94/24 | NC_017371.1 | 5 | 2 |
| SouthAfrica20 | NC_022130.1 | 4 | 1 |
| India7 | NC_017372.1 | 4 | 1 |
| BM012A | NC_022886.1 | 5 | 4 |
| BM012S | NC_022911.1 | 4 | 4 |
| BM012B | NZ_CP007605.1 | 4 | 4 |
| Strains | Region | IS605 | IS606 | IS607 | IS608 | IS609 |
|---|---|---|---|---|---|---|
| NY40 | East Asia | 1 | 4 | 1 | 0 | 0 |
| ML3 | East Asia | 0 | 1 | 0 | 0 | 0 |
| UM032, UM298, UM299, F30, F57, ML1, UM066, OK310, 52, Hp238 | East Asia | 0 | 0 | 0 | 0 | 0 |
| UM037 | East Asia | 5 | 0 | 0 | 0 | 0 |
| F32 | East Asia | 1 | 0 | 0 | 0 | 0 |
| XZ274 | East Asia | 0 | 0 | 2 | 0 | 0 |
| F16, OK113 | East Asia | 0 | 0 | 1 | 0 | 0 |
| oki128, oki154, oki673, oki828 | East Asia | 0 | 3 | 0 | 0 | 0 |
| oki102, oki112, oki422, oki898 | East Asia | 0 | 0 | 0 | 0.5 | 1 |
| 26695-1CL, 26695-1CH, 26695-1 | East Asia | 5 | 2 | 0 | 0 | 0 |
| Shi112 | South America | 0 | 0 | 5 | 2 | 0 |
| Sat464 | South America | 0.5 | 0.5 | 1 | 0 | 0 |
| Cuz20 | South America | 0 | 0.5 | 1 | 1 | 0 |
| PeCan4 | South America | 0 | 0.5 | 0 | 1 | 0 |
| PeCan18 | South America | 0 | 0 | 0 | 2 | 0 |
| Puno120 | South America | 0 | 0 | 0 | 0 | 0 |
| Shi169 | South America | 0 | 0.5 | 6 | 0 | 0 |
| SJM180 | South America | 0 | 0 | 0 | 0 | 1 |
| Puno135, Shi417 | South America | 0 | 0.5 | 0 | 0 | 0 |
| v225d | South America | 1 | 0.5 | 0 | 0 | 0 |
| 7C, J166 | North America | 0 | 0 | 0 | 0 | 0.5 |
| 29CaP | North America | 0 | 1 | 0 | 0 | 4 |
| Aklavik117 | North America | 0 | 0.5 | 1 | 0 | 0 |
| 26695-1, 26695-1MET | North America | 5 | 2 | 0 | 0 | 0 |
| J99 | North America | 0 | 1 | 0 | 0 | 0.5 |
| ELS37 | North America | 0 | 6 | 1 | 0 | 0 |
| B38 | Europe | 0 | 0 | 0 | 0 | 5 |
| HUP-B14 | Europe | 0 | 0 | 0 | 1 | 1 |
| Rif1, Rif2, 26,695 | Europe | 5 | 2 | 0 | 0 | 0 |
| B8 | Europe | 0 | 0 | 0 | 1 | 0.5 |
| G27 | Europe | 5 | 0 | 0 | 0 | 0 |
| Lithuania75, P12 | Europe | 0 | 0 | 0 | 0 | 0 |
| 2017, 2018, 908 | Europe | 0 | 1 | 0 | 0 | 0.5 |
| SouthAfrica7 | Africa | 0 | 2 | 0 | 0 | 0.5 |
| Gambia94/24, SouthAfrica20 | Africa | 0 | 0 | 0 | 0 | 0.5 |
| India7, Santal49 | India | 0 | 0 | 0 | 0 | 0 |
| BM012A, BM012B, BM012S | Australia | 0 | 0 | 9 | 0.5 | 0 |
| BM013A, BM013B | Australia | 0 | 0 | 4 | 0 | 0 |
| 83 | Unknown | 1 | 0 | 0 | 0 | 0 |
| 35A | unknown | 0 | 0 | 0 | 0 | 0 |
| IS | World-Wide BPs | Region-Specific BPs | Strain-Specific BPs |
|---|---|---|---|
| IS605 | 4 | 0 | 1 |
| IS606 | 3 | 1 | 0 |
| IS607 | 0 | 3 | 0 |
| IS608 | 0 | 1 | 0 |
| IS609 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noureen, M.; Kawashima, T.; Arita, M. Genetic Markers of Genome Rearrangements in Helicobacter pylori. Microorganisms 2021, 9, 621. https://doi.org/10.3390/microorganisms9030621
Noureen M, Kawashima T, Arita M. Genetic Markers of Genome Rearrangements in Helicobacter pylori. Microorganisms. 2021; 9(3):621. https://doi.org/10.3390/microorganisms9030621
Chicago/Turabian StyleNoureen, Mehwish, Takeshi Kawashima, and Masanori Arita. 2021. "Genetic Markers of Genome Rearrangements in Helicobacter pylori" Microorganisms 9, no. 3: 621. https://doi.org/10.3390/microorganisms9030621
APA StyleNoureen, M., Kawashima, T., & Arita, M. (2021). Genetic Markers of Genome Rearrangements in Helicobacter pylori. Microorganisms, 9(3), 621. https://doi.org/10.3390/microorganisms9030621