
Hepatitis B Virus Genotype Study in West Africa Reveals an Expanding Clade of Subgenotype A4

 ,
,  , , ,
, , , 

Abstract
1. Introduction
2. Materials and Methods
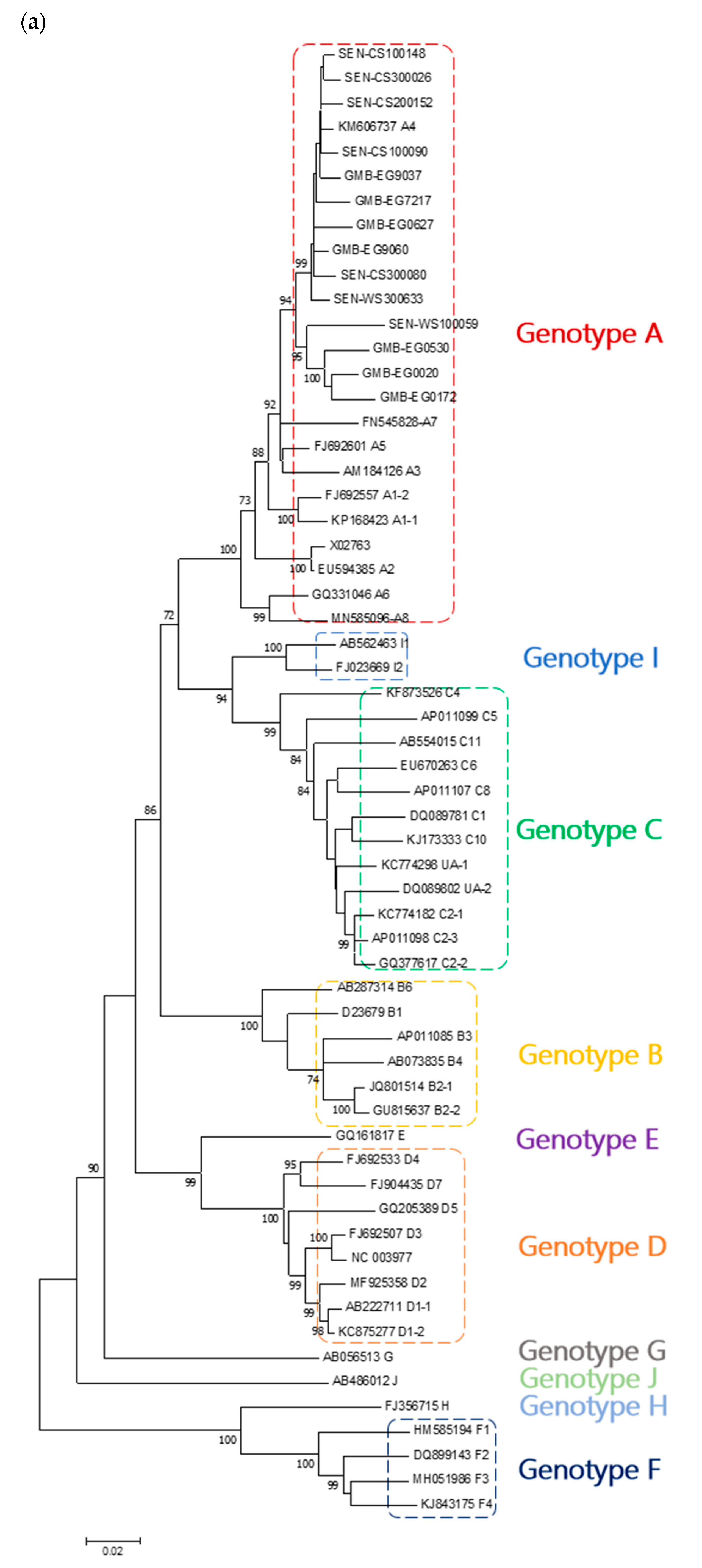
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Norder, H.; Hammas, B.; Lee, S.D.; Bile, K.; Courouce, A.M.; Mushahwar, I.K.; Magnius, L.O. Genetic relatedness of hepatitis B viral strains of diverse geographical origin and natural variations in the primary structure of the surface antigen. J. Gen. Virol. 1993, 74, 1341–1348. [Google Scholar] [CrossRef]
- Stuyver, L.; De Gendt, S.; Van Geyt, C.; Zoulim, F.; Fried, M.; Schinazi, R.F.; Rossau, R. A new genotype of hepatitis B virus: Complete genome and phylogenetic relatedness. J. Gen. Virol. 2000, 81, 67–74. [Google Scholar] [CrossRef]
- Norder, H.; Couroucé, A.M.; Coursaget, P.; Echevarria, J.M.; Lee, S.D.; Mushahwar, I.K.; Robertson, B.H.; Locarnini, S.; Magnius, L.O. Genetic diversity of hepatitis B virus strains derived worldwide: Genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004, 47, 289–309. [Google Scholar] [CrossRef]
- Kramvis, A.; Kew, M.C. Relationship of genotypes of hepatitis B virus to mutations, disease progression and response to antiviral therapy. J. Viral Hepat. 2005, 12, 456–464. [Google Scholar] [CrossRef]
- Sugauchi, F.; Orito, E.; Kato, H.; Suzuki, S.; Kawakita, S.; Sakamoto, Y.; Fukushima, K.; Akiba, T.; Yoshihara, N.; Ueda, R.; et al. Genotype, serotype, and phylogenetic characterization of the complete genome sequence of hepatitis B virus isolates from Malawian chronic carriers of the virus. J. Med. Virol. 2003, 69, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A.; Arakawa, K.; Yu, M.C.; Nogueira, R.; Stram, D.O.; Kew, M.C. Relationship of serological subtype, basic core promoter and precore mutations to genotypes/subgenotypes of hepatitis B virus. J. Med. Virol. 2008, 80, 27–46. [Google Scholar] [CrossRef]
- Rajoriya, N.; Combet, C.; Zoulim, F.; Janssen, H.L.A. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017, 67, 1281–1297. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, C.L.; Aguilar, J.C.; Aguiar, J.; Muzio, V.; Pentón, E.; Garcia, D.; Guillen, G.; Pujol, F.H. HBV genotypic variability in Cuba. PLoS ONE 2015, 10, e118959. [Google Scholar] [CrossRef]
- Sugauchi, F.; Kumada, H.; Acharya, S.A.; Shrestha, S.M.; Gamutan, M.T.A.; Khan, M.; Gish, R.G.; Tanaka, Y.; Kato, T.; Orito, E.; et al. Epidemiological and sequence differences between two subtypes (Ae and Aa) of hepatitis B virus genotype A. J. Gen. Virol. 2004, 85, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, S.M.; Van Staden, L.; Kew, M.C.; Sim, J.G.M. A unique segment of the hepatitis B virus group A genotype identified in isolates from South Africa. J. Gen. Virol. 1997, 78, 1719–1729. [Google Scholar] [CrossRef]
- Kramvis, A.; Weitzmann, L.; Owiredu, W.K.B.A.; Kew, M.C. Analysis of the complete genome of subgroup A′ hepatitis B virus isolates from South Africa. J. Gen. Virol. 2002, 83, 835–839. [Google Scholar] [CrossRef]
- Kimbi, G.C.; Kramvis, A.; Kew, M.C. Distinctive sequence characteristics of subgenotype A1 isolates of hepatitis B virus from South Africa. J. Gen. Virol. 2004, 85, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Kurbanov, F.; Tanaka, Y.; Fujiwara, K.; Sugauchi, F.; Mbanya, D.; Zekeng, L.; Ndembi, N.; Ngansop, C.; Kaptue, L.; Miura, T.; et al. A new subtype (subgenotype) Ac (A3) of hepatitis B virus and recombination between genotypes A and E in Cameroon. J. Gen. Virol. 2005, 86, 2047–2056. [Google Scholar] [CrossRef]
- Olinger, C.M.; Venard, V.; Njayou, M.; Bola Oyefolu, A.O.; Maïga, I.; Kemp, A.J.; Omilabu, S.A.; le Faou, A.; Muller, C.P. Phylogenetic analysis of the precore/core gene of hepatitis B virus genotypes E and A in West Africa: New subtypes, mixed infections and recombinations. J. Gen. Virol. 2006, 87, 1163–1173. [Google Scholar] [CrossRef]
- Andernach, I.E.; Nolte, C.; Pape, J.W.; Muller, C.P. Slave trade and hepatitis B virus genotypes and subgenotypes in Haiti and Africa. Emerg. Infect. Dis. 2009, 15, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Pourkarim, M.R.; Lemey, P.; Amini-Bavil-Olyaee, S.; Maes, P.; Van Ranst, M. Novel hepatitis B virus subgenotype A6 in African-Belgian patients. J. Clin. Virol. 2010, 47, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Hübschen, J.M.; Mbah, P.O.; Forbi, J.C.; Otegbayo, J.A.; Olinger, C.M.; Charpentier, E.; Muller, C.P. Detection of a new subgenotype of hepatitis B virus genotype A in Cameroon but not in neighbouring Nigeria. Clin. Microbiol. Infect. 2011, 17, 88–94. [Google Scholar] [CrossRef]
- Thijssen, M.; Trovão, N.S.; Mina, T.; Maes, P.; Pourkarim, M.R. Novel hepatitis B virus subgenotype A8 and quasi-subgenotype D12 in African–Belgian chronic carriers. Int. J. Infect. Dis. 2020, 93, 98–101. [Google Scholar] [CrossRef]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Kurbanov, F.; Van Ranst, M.; Tacke, F. Molecular identification of hepatitis B virus genotypes/ subgenotypes: Revised classification hurdles and updated resolutions. World J. Gastroenterol. 2014, 20, 7152–7168. [Google Scholar] [CrossRef]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Lemey, P.; Maes, P.; Van Ranst, M. Are hepatitis B virus “subgenotypes” defined accurately? J. Clin. Virol. 2010, 47, 356–360. [Google Scholar] [CrossRef]
- Pourkarim, M.R.; Amini-Bavil-Olyaee, S.; Lemey, P.; Maes, P.; Ranst, M. Van HBV subgenotype misclassification expands quasi-subgenotype A3. Clin. Microbiol. Infect. 2011, 17, 947–949. [Google Scholar] [CrossRef]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- McNaughton, A.L.; Revill, P.A.; Littlejohn, M.; Matthews, P.C.; Azim Ansari, M. Analysis of genomic-length HBV sequences to determine genotype and subgenotype reference sequences. J. Gen. Virol. 2020, 101, 271–283. [Google Scholar] [CrossRef]
- Cohen, D.; Ghosh, S.; Shimakawa, Y.; Ramou, N.; Garcia, P.S.; Dubois, A.; Guillot, C.; Kakwata-Nkor Deluce, N.; Tilloy, V.; Durand, G.; et al. HBV PreS2Δ38-55 variants: A newly identified risk factor for hepatocellular carcinoma. JHEP Rep. 2020, 2, 100144. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Li, B.C.; Miska, S.; Krüger, D.H.; Meisel, H.; Will, H. A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J. Virol. 1995, 69, 5437–5444. [Google Scholar] [CrossRef]
- Günther, S.; Sommer, G.; Von Breunig, F.; Iwanska, A.; Kalinina, T.; Sterneck, M.; Will, H. Amplification of full-length hepatitis B virus genomes from samples from patients with low levels of viremia: Frequency and functional consequences of PCR-introduced mutations. J. Clin. Microbiol. 1998, 36, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Bulla, I.; Abdou-chekaraou, M.; Gordien, E.; Stanke, M.; Morgenstern, B.; Zoulim, F.; De, P. jpHMM: Recombination analysis in viruses with circular genomes such as the hepatitis B virus. Nucleic Acids Res. 2012, 40, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Liebert, M.A.; Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar]
- Purdy, M.A. Hepatitis B virus S gene escape mutants. Asian J. Transfus. Sci. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Toyé, R.M.; Lô, G.; Diop-Ndiaye, H.; Cissé, A.M.; Ndiaye, A.J.S.; Kébé-Fall, K.; Dramé, A.; Cohen, D.; Pujol, F.H.; Mboup, S.; et al. Prevalence and molecular characterization of hepatitis B virus infection in HIV-infected children in Senegal. Clin. Res. Hepatol. Gastroenterol. 2020. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Subgenotype | Expanding Clade A4 |
|---|---|
| A1 | 5.3 ± 0.004 |
| A2 | 5.4 ± 0.003 |
| A3 | 5.0 ± 0.004 |
| A4 | 3.9 ± 0.004 |
| A5 | 4.2 ± 0.004 |
| A6 | 5.5 ± 0.004 |
| A7 | 5.6 ± 0.006 |
| A8 | 6.4 ± 0.004 |
| Expanding clade A4 | (2.9 ± 0.002) |
| Amino Acid Position in the S Surface Gene | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample | Genotype | Serotype | 122 | 160 | 127 | 159 | 140 | or 134 | |
| 1 | CS100090 | A4 | ayw1 | R | K | P | A | T | F |
| 2 | CS100148 | A4 | ayw1 | R | K | P | A | T | F |
| 3 | CS200152 | A4 | ayw1 | R | K | P | A | T | F |
| 4 | CS300026 | A4 | ayw1 | R | K | P | A | T | I |
| 5 | CS300080 | A4 | ayw1/2 | R | K | P | G | T | F |
| 6 | EG0020 | New clade A4 | ayw1/2 | R | K | P | G | T | F |
| 7 | EG0172 | New clade A4 | ayw1/2 | R | K | P | G | T | F |
| 8 | EG0530 | New clade A4 | ayw1/2 | R | K | P | G | T | F |
| 9 | EG0627 | A4 | ayw1 | R | K | P | A | T | F |
| 10 | EG7217 | A4 | ayw1 | R | K | P | A | T | F |
| 11 | EG9037 | A4 | ayw1 | R | K | P | A | T | F |
| 12 | EG9060 | A4 | ayw1 | R | K | P | A | T | F |
| 13 | WS100059 | Close to clade A4 | ayw1/2 | R | K | P | G | T | F |
| 14 | WS300633 | A4 | ayw1 | R | K | P | A | T | F |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toyé, R.M.; Cohen, D.; Pujol, F.H.; Sow-Sall, A.; Lô, G.; Hoshino, K.; Mizokami, M.; Zoulim, F.; Lemoine, M.; Touré-Kane, C.; et al. Hepatitis B Virus Genotype Study in West Africa Reveals an Expanding Clade of Subgenotype A4. Microorganisms 2021, 9, 623. https://doi.org/10.3390/microorganisms9030623
Toyé RM, Cohen D, Pujol FH, Sow-Sall A, Lô G, Hoshino K, Mizokami M, Zoulim F, Lemoine M, Touré-Kane C, et al. Hepatitis B Virus Genotype Study in West Africa Reveals an Expanding Clade of Subgenotype A4. Microorganisms. 2021; 9(3):623. https://doi.org/10.3390/microorganisms9030623
Chicago/Turabian StyleToyé, Rayana Maryse, Damien Cohen, Flor Helene Pujol, Amina Sow-Sall, Gora Lô, Kunikazu Hoshino, Masashi Mizokami, Fabien Zoulim, Maud Lemoine, Coumba Touré-Kane, and et al. 2021. "Hepatitis B Virus Genotype Study in West Africa Reveals an Expanding Clade of Subgenotype A4" Microorganisms 9, no. 3: 623. https://doi.org/10.3390/microorganisms9030623
APA StyleToyé, R. M., Cohen, D., Pujol, F. H., Sow-Sall, A., Lô, G., Hoshino, K., Mizokami, M., Zoulim, F., Lemoine, M., Touré-Kane, C., & Chemin, I. (2021). Hepatitis B Virus Genotype Study in West Africa Reveals an Expanding Clade of Subgenotype A4. Microorganisms, 9(3), 623. https://doi.org/10.3390/microorganisms9030623