
The Ubiquitination System within Bacterial Host–Pathogen Interactions


Abstract
:1. Introduction
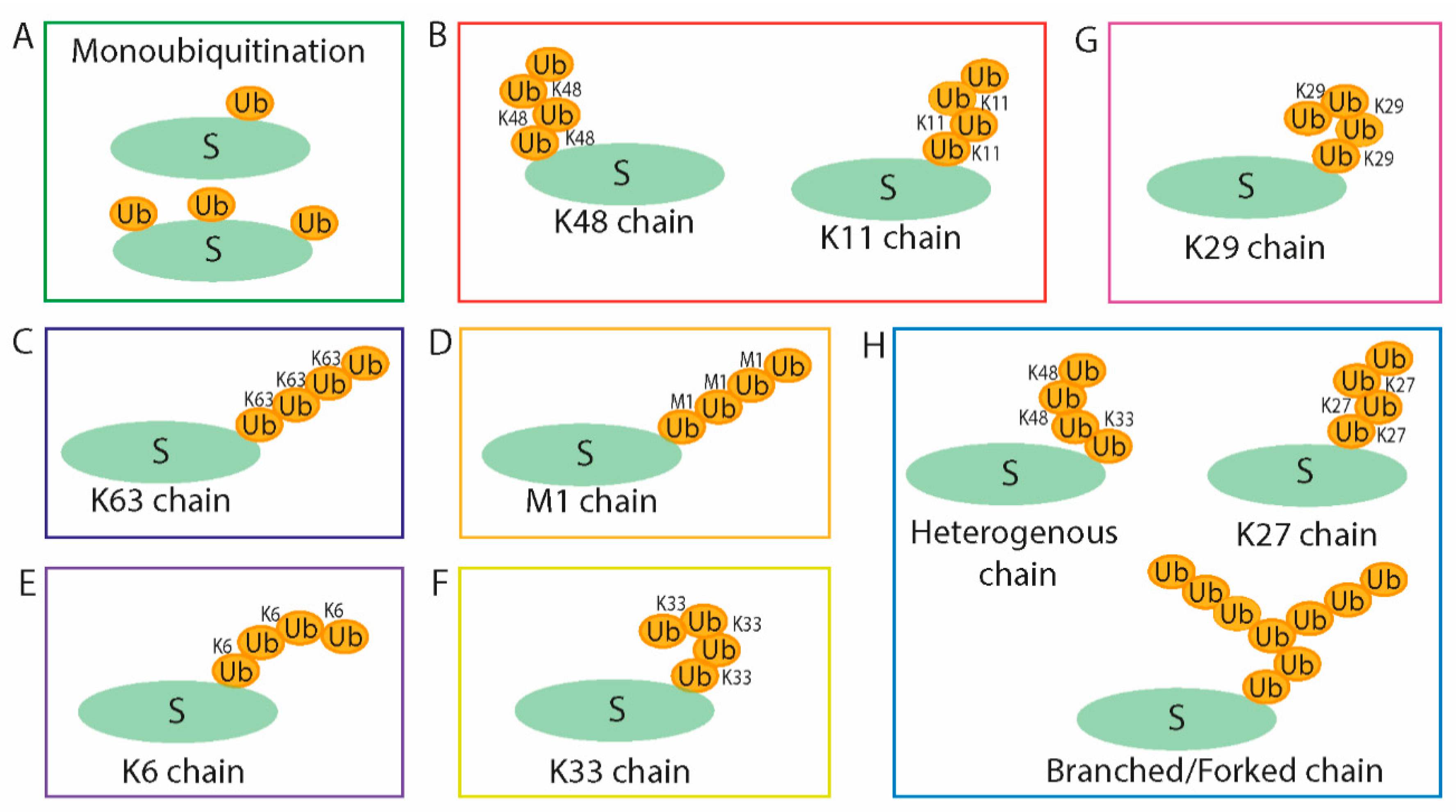
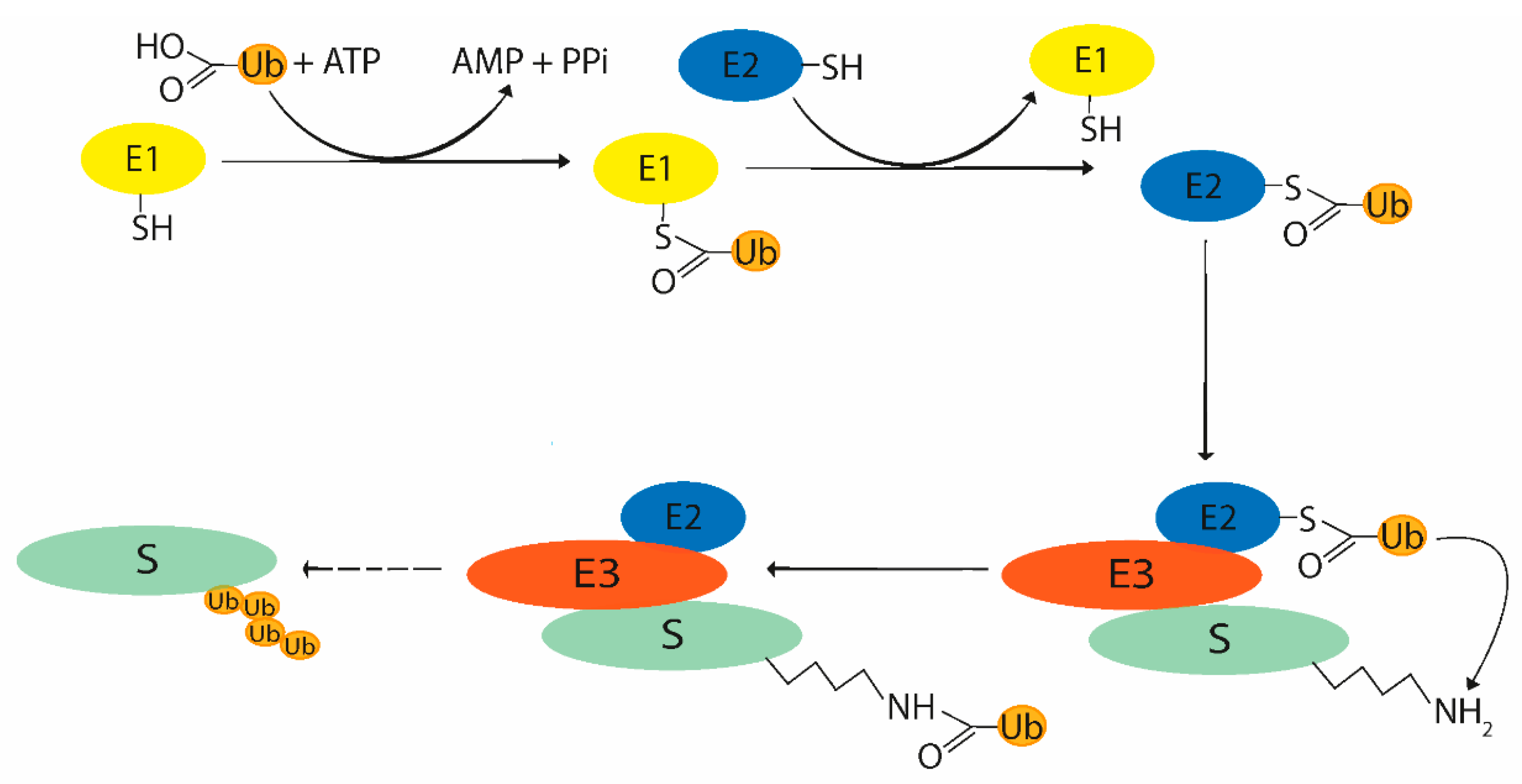
2. Ubiquitin System
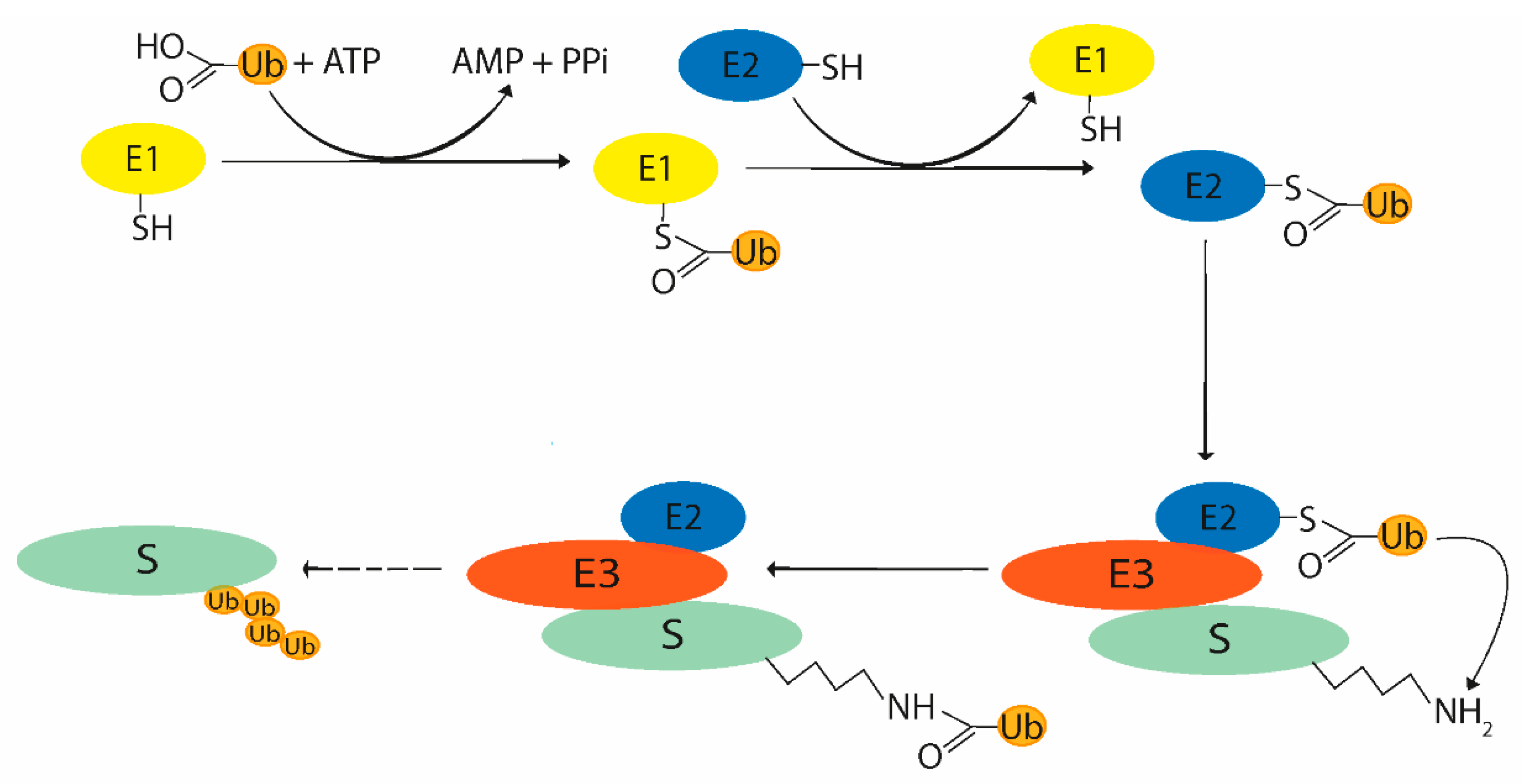
2.1. Ubiquitination Process
2.2. Enzymes Participating in Ubiquitin System
2.3. Ubiquitin Ligases
2.4. Deubiquitinating Enzymes
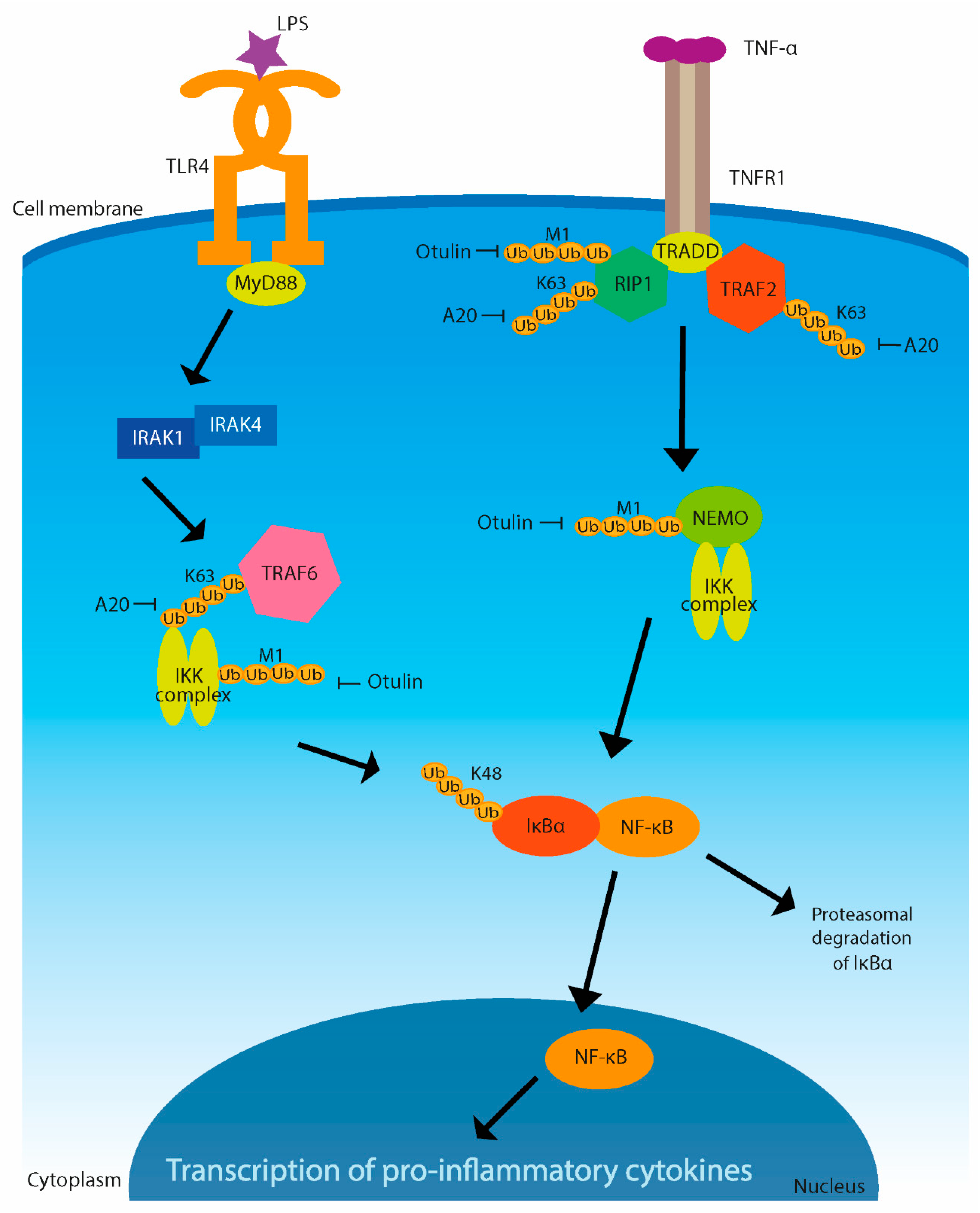
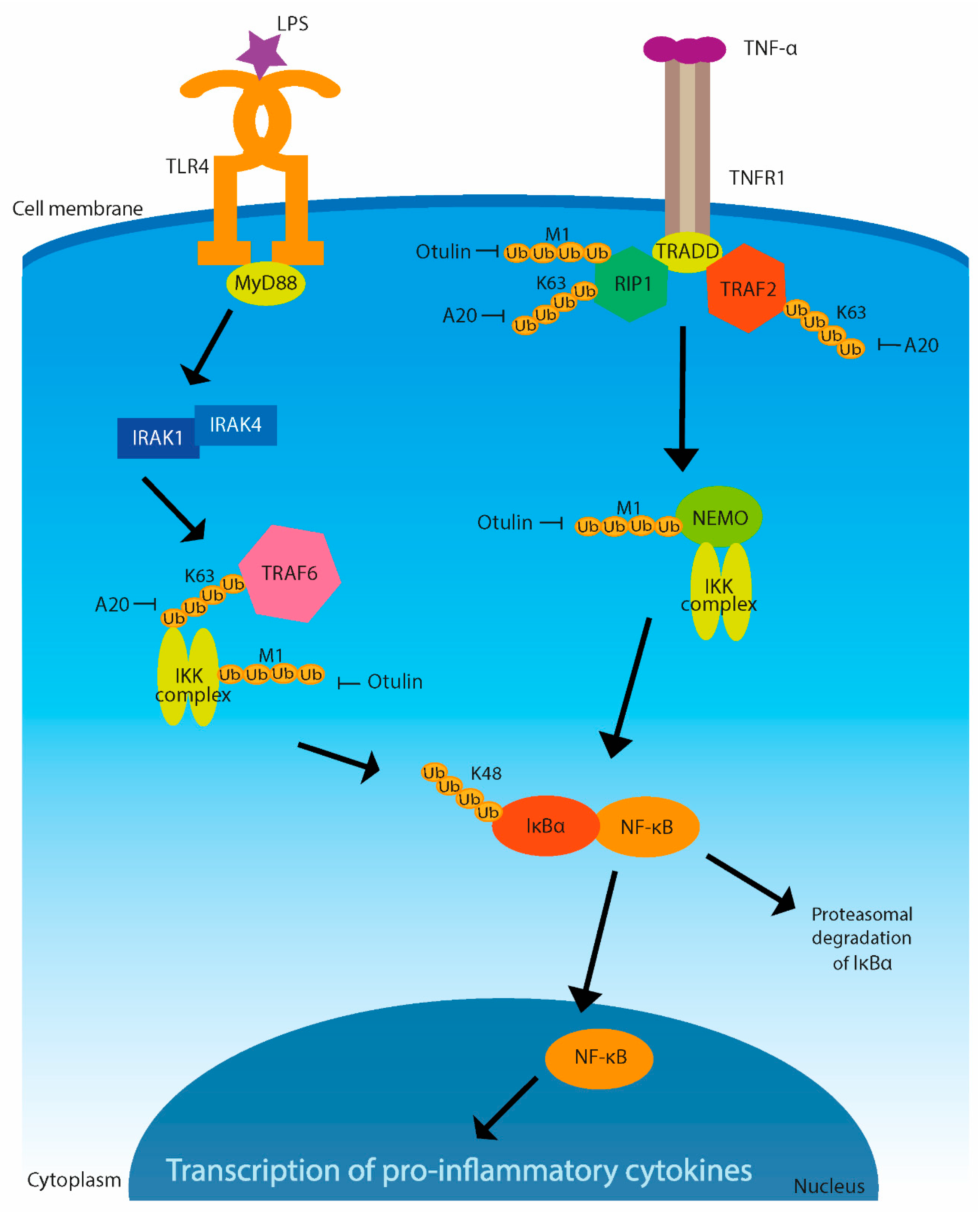
3. Immune Signaling Regulated by Ubiquitination
4. Host Ubiquitination Machinery as a Target for Pathogens
4.1. Bacterial Influence on Host Ubiquitinating and Deubiquitinating Processes
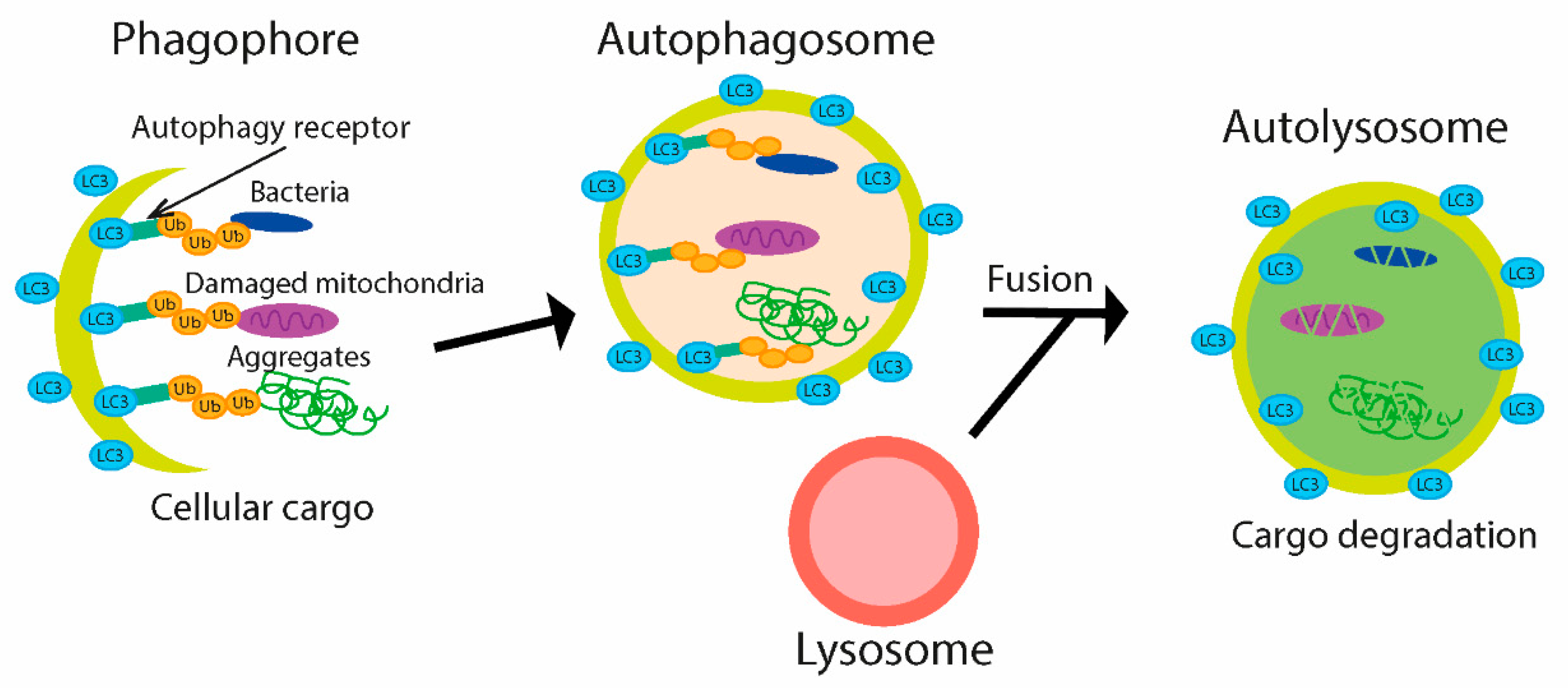
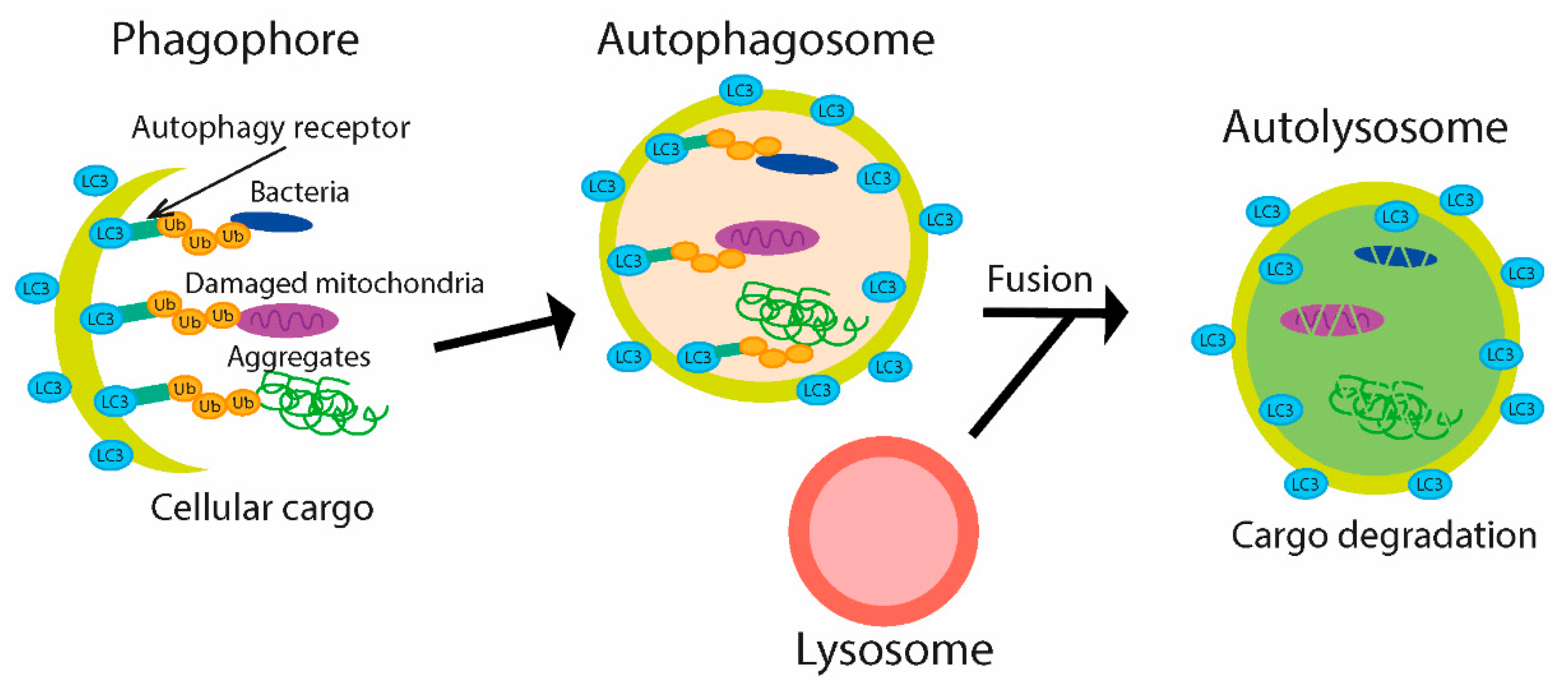
4.2. Bacterial Modulation of Ubiquitin-Mediated Autophagy during Infection
4.3. Bacterial Effectors Intervene with Ubiquitination
4.3.1. The Effect of Yersinia on the Host Immune Response
4.3.2. Salmonella Produces Various Effectors
4.3.3. Escherichia Effectors
4.3.4. Miscellaneous Protein Effectors of Legionella
4.3.5. Insidious Shigella Effectors
4.3.6. Chlamydia DUB Effectors
5. Ubiquitination and DUBs in Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, Functions, Mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1695, 55–72. [Google Scholar] [CrossRef] [Green Version]
- Nijman, S.M.B.; Luna-Vargas, M.P.A.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.G.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Haglund, K.; Dikic, I. Ubiquitylation and Cell Signaling. EMBO J. 2005, 24, 3353–3359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.-W.; Ryu, K.-Y. Cellular Ubiquitin Pool Dynamics and Homeostasis. BMB Rep. 2014, 47, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the Polyubiquitin Proteolytic Signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative Proteomics Reveals the Function of Unconventional Ubiquitin Chains in Proteasomal Degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.T.; D’Andrea, A.D. Regulation of DNA Repair by Ubiquitylation. Nat. Rev. Mol. Cell Biol. 2006, 7, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.-C.; Lee, Y.-R.; Lin, S.-Y.; Chang, L.-Y.; Tan, Y.P.; Hung, C.-C.; Kuo, J.-C.; Liu, C.-H.; Lin, M.-Y.; Xu, M.; et al. K33-Linked Polyubiquitination of Coronin 7 by Cul3-KLHL20 Ubiquitin E3 Ligase Regulates Protein Trafficking. Mol. Cell 2014, 54, 586–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, A.E.H.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; Everley, R.A.; Dephoure, N.; Zhou, C.; Koren, I.; Gygi, S.P.; Elledge, S.J. Quantitative Proteomic Atlas of Ubiquitination and Acetylation in the DNA Damage Response. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.R.; Solomon, E. BRCA1: BARD1 Induces the Formation of Conjugated Ubiquitin Structures, Dependent on K6 of Ubiquitin, in Cells during DNA Replication and Repair. Hum. Mol. Genet. 2004, 13, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Manzanillo, P.S.; Ayres, J.S.; Watson, R.O.; Collins, A.C.; Souza, G.; Rae, C.S.; Schneider, D.S.; Nakamura, K.; Shiloh, M.U.; Cox, J.S. The Ubiquitin Ligase Parkin Mediates Resistance to Intracellular Pathogens. Nature 2013, 501, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [Green Version]
- Besche, H.C.; Sha, Z.; Kukushkin, N.V.; Peth, A.; Hock, E.-M.; Kim, W.; Gygi, S.; Gutierrez, J.A.; Liao, H.; Dick, L.; et al. Autoubiquitination of the 26S Proteasome on Rpn13 Regulates Breakdown of Ubiquitin Conjugates. EMBO J. 2014, 33, 1159–1176. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and Quantitative Assessment of the Ubiquitin-Modified Proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Xie, X.; Xiao, Y.; Hu, H.; Zou, Q.; Cheng, X.; Sun, S.-C. Epigenetic Regulation of the Expression of Il12 and Il23 and Autoimmune Inflammation by the Deubiquitinase Trabid. Nat. Immunol. 2016, 17, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Hamada, F.; Schwarz-Romond, T.; Bienz, M. Trabid, a New Positive Regulator of Wnt-Induced Transcription with Preference for Binding and Cleaving K63-Linked Ubiquitin Chains. Genes Dev. 2008, 22, 528–542. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of Linear Polyubiquitylation of NEMO in NF-KappaB Activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.S. Protein Modification by SUMO. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef] [Green Version]
- Ribet, D.; Cossart, P. Ubiquitin, SUMO, and NEDD8: Key Targets of Bacterial Pathogens. Trends Cell Biol. 2018, 28, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Panasenko, O.O. Identification of Ubiquitinated Proteins. Mater. Methods 2014, 4, 827. [Google Scholar] [CrossRef]
- Mulder, M.P.C.; Witting, K.; Berlin, I.; Pruneda, J.N.; Wu, K.-P.; Chang, J.-G.; Merkx, R.; Bialas, J.; Groettrup, M.; Vertegaal, A.C.O.; et al. A Cascading Activity-Based Probe Sequentially Targets E1–E2–E3 Ubiquitin Enzymes. Nat. Chem. Biol. 2016, 12, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Neutzner, M.; Neutzner, A. Enzymes of Ubiquitination and Deubiquitination. Essays Biochem. 2012, 52, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Rape, M. Building Ubiquitin Chains: E2 Enzymes at Work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Ebner, P.; Versteeg, G.A.; Ikeda, F. Ubiquitin Enzymes in the Regulation of Immune Responses. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 425–460. [Google Scholar] [CrossRef]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-Type E3 Ligases: Master Manipulators of E2 Ubiquitin-Conjugating Enzymes and Ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING Finger Families of E3 Ubiquitin Ligases at a Glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Kumar, S. Mammalian HECT Ubiquitin-Protein Ligases: Biological and Pathophysiological Aspects. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.J.; Sixma, T.K. “Ubiquitylation: Mechanism and Functions” Review Series: RBR E3-Ligases at Work. EMBO Rep. 2014, 15, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 Ubiquitin Ligases: New Structures, New Insights, New Questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef] [Green Version]
- Lechtenberg, B.C.; Rajput, A.; Sanishvili, R.; Dobaczewska, M.K.; Ware, C.F.; Mace, P.D.; Riedl, S.J. Structure of a HOIP/E2~ubiquitin Complex Reveals RBR E3 Ligase Mechanism and Regulation. Nature 2016, 529, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, K.D. DUBs at a Glance. J. Cell Sci. 2009, 122, 2325–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.-Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwasna, D.; Abdul Rehman, S.A.; Natarajan, J.; Matthews, S.; Madden, R.; De Cesare, V.; Weidlich, S.; Virdee, S.; Ahel, I.; Gibbs-Seymour, I.; et al. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol. Cell 2018, 70, 150–164.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komander, D.; Clague, M.J.; Urbé, S. Breaking the Chains: Structure and Function of the Deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Hu, H.; Sun, S.-C. Ubiquitin Signaling in Immune Responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-Like Receptor and RIG-1-Like Receptor Signaling. Available online: https://nyaspubs.onlinelibrary.wiley.com/doi/abs/10.1196/annals.1443.020 (accessed on 23 August 2019).
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the Immune System. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Dynek, J.N.; Zobel, K.; Deshayes, K.; Fairbrother, W.J.; Vucic, D. C-IAP1 and c-IAP2 Are Critical Mediators of Tumor Necrosis Factor α (TNFα)-Induced NF-ΚB Activation. J. Biol. Chem. 2008, 283, 24295–24299. [Google Scholar] [CrossRef] [Green Version]
- Dynek, J.N.; Goncharov, T.; Dueber, E.C.; Fedorova, A.V.; Izrael-Tomasevic, A.; Phu, L.; Helgason, E.; Fairbrother, W.J.; Deshayes, K.; Kirkpatrick, D.S.; et al. C-IAP1 and UbcH5 Promote K11-Linked Polyubiquitination of RIP1 in TNF Signalling. EMBO J. 2010, 29, 4198–4209. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Goncharov, T.; Maecker, H.; Zobel, K.; Kömüves, L.G.; Deshayes, K.; Vucic, D. Cellular Inhibitors of Apoptosis Are Global Regulators of NF-ΚB and MAPK Activation by Members of the TNF Family of Receptors. Sci. Signal. 2012, 5, ra22. [Google Scholar] [CrossRef]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN Antagonizes LUBAC Signaling by Specifically Hydrolyzing Met1-Linked Polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-Ubiquitination and Ubiquitin Ligase Domains of A20 Downregulate NF-ΚB Signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Conze, D.B.; Wu, C.-J.; Thomas, J.A.; Landstrom, A.; Ashwell, J.D. Lys63-Linked Polyubiquitination of IRAK-1 Is Required for Interleukin-1 Receptor- and Toll-like Receptor-Mediated NF-KappaB Activation. Mol. Cell. Biol. 2008, 28, 3538–3547. [Google Scholar] [CrossRef] [Green Version]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD Proteins: Regulators of Inflammation in Health and Disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef]
- Hitotsumatsu, O.; Ahmad, R.-C.; Tavares, R.; Wang, M.; Philpott, D.; Turer, E.E.; Lee, B.L.; Shiffin, N.; Advincula, R.; Malynn, B.A.; et al. The Ubiquitin-Editing Enzyme A20 Restricts Nucleotide-Binding Oligomerization Domain Containing 2-Triggered Signals. Immunity 2008, 28, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.R.; Levy, O. 3—Innate Immunity. In Clinical Immunology, 5th ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Elsevier: London, UK, 2019; pp. 39–53. ISBN 978-0-7020-6896-6. [Google Scholar]
- Xiao, Y.; Huang, Q.; Wu, Z.; Chen, W. Roles of Protein Ubiquitination in Inflammatory Bowel Disease. Immunobiology 2020, 225, 152026. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.; Jobin, C. Ubiquitin Protein Modification and Signal Transduction: Implications for Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2005, 11, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Chatzidaki-Livanis, M.; Coyne, M.J.; Roelofs, K.G.; Gentyala, R.R.; Caldwell, J.M.; Comstock, L.E. Gut Symbiont Bacteroides Fragilis Secretes a Eukaryotic-Like Ubiquitin Protein That Mediates Intraspecies Antagonism. mBio 2017, 8, e01902-17. [Google Scholar] [CrossRef] [Green Version]
- Rytkönen, A.; Holden, D.W. Bacterial Interference of Ubiquitination and Deubiquitination. Cell Host Microbe 2007, 1, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruneda, J.N.; Durkin, C.H.; Geurink, P.P.; Ovaa, H.; Santhanam, B.; Holden, D.W.; Komander, D. The Molecular Basis for Ubiquitin and Ubiquitin-like Specificities in Bacterial Effector Proteases. Mol. Cell 2016, 63, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, L.A.; Edelmann, M.J. Ubiquitination as an Efficient Molecular Strategy Employed in Salmonella Infection. Front. Immunol. 2014, 5, 558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronau, J.A.; Beckmann, J.F.; Hochstrasser, M. Substrate Specificity of the Ubiquitin and Ubl Proteases. Cell Res. 2016, 26, 441–456. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, M.J.; Kramer, H.B.; Altun, M.; Kessler, B.M. Post-Translational Modification of the Deubiquitinating Enzyme Otubain 1 Modulates Active RhoA Levels and Susceptibility to Yersinia Invasion. FEBS J. 2010, 277, 2515–2530. [Google Scholar] [CrossRef]
- Mukherjee, S.; Keitany, G.; Li, Y.; Wang, Y.; Ball, H.L.; Goldsmith, E.J.; Orth, K. Yersinia YopJ Acetylates and Inhibits Kinase Activation by Blocking Phosphorylation. Science 2006, 312, 1211–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Monack, D.M.; Kayagaki, N.; Wertz, I.; Yin, J.; Wolf, B.; Dixit, V.M. Yersinia Virulence Factor YopJ Acts as a Deubiquitinase to Inhibit NF-ΚB Activation. J. Exp. Med. 2005, 202, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Higashide, W.M.; McCormick, B.A.; Chen, J.; Zhou, D. The Inflammation-Associated Salmonella SopA Is a HECT-like E3 Ubiquitin Ligase. Mol. Microbiol. 2006, 62, 786–793. [Google Scholar] [CrossRef]
- Negrate, G.L.; Faustin, B.; Welsh, K.; Loeffler, M.; Krajewska, M.; Hasegawa, P.; Mukherjee, S.; Orth, K.; Krajewski, S.; Godzik, A.; et al. Salmonella Secreted Factor L Deubiquitinase of Salmonella Typhimurium Inhibits NF-ΚB, Suppresses IκBα Ubiquitination and Modulates Innate Immune Responses. J. Immunol. 2008, 180, 5045–5056. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, F.S.; Thomas, M.; Sachse, M.; Santos, A.J.M.; Figueira, R.; Holden, D.W. The Salmonella Deubiquitinase SseL Inhibits Selective Autophagy of Cytosolic Aggregates. PLoS Pathog. 2012, 8, e1002743. [Google Scholar] [CrossRef]
- Ye, Z.; Petrof, E.O.; Boone, D.; Claud, E.C.; Sun, J. Salmonella Effector AvrA Regulation of Colonic Epithelial Cell Inflammation by Deubiquitination. Am. J. Pathol. 2007, 171, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Sirisaengtaksin, N.; O’Donoghue, E.J.; Jabbari, S.; Roe, A.J.; Krachler, A.M. Bacterial Outer Membrane Vesicles Provide an Alternative Pathway for Trafficking of Type III Secreted Effectors into Epithelial Cells. bioRxiv 2018, 415794. [Google Scholar] [CrossRef]
- Bhogaraju, S.; Kalayil, S.; Liu, Y.; Bonn, F.; Colby, T.; Matic, I.; Dikic, I. Phosphoribosylation of Ubiquitin Promotes Serine Ubiquitination and Impairs Conventional Ubiquitination. Cell 2016, 167, 1636–1649.e13. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Liu, Y.; Gonzalez, A.; Bonn, F.; Liu, Y.; Rogov, V.V.; Heinz, M.; Stolz, A.; Hummer, G.; et al. Regulation of Phosphoribosyl-Linked Serine Ubiquitination by Deubiquitinases DupA and DupB. Mol. Cell 2020, 77, 164–179.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheedlo, M.J.; Qiu, J.; Tan, Y.; Paul, L.N.; Luo, Z.-Q.; Das, C. Structural Basis of Substrate Recognition by a Bacterial Deubiquitinase Important for Dynamics of Phagosome Ubiquitination. Proc. Natl. Acad. Sci. USA 2015, 112, 15090–15095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubori, T.; Kitao, T.; Ando, H.; Nagai, H. LotA, a Legionella Deubiquitinase, Has Dual Catalytic Activity and Contributes to Intracellular Growth. Cell. Microbiol. 2018, 20, e12840. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Wang, X.; Huang, C.; Xu, D.; Wang, Z.; Zhou, Y.; Zhu, Y. A Bacterial Effector Deubiquitinase Specifically Hydrolyses Linear Ubiquitin Chains to Inhibit Host Inflammatory Signalling. Nat. Microbiol. 2019, 4, 1282–1293. [Google Scholar] [CrossRef]
- Kubori, T.; Bui, X.T.; Hubber, A.; Nagai, H. Legionella RavZ Plays a Role in Preventing Ubiquitin Recruitment to Bacteria-Containing Vacuoles. Front. Cell. Infect. Microbiol. 2017, 7, 384. [Google Scholar] [CrossRef]
- Nakagawa, I. Streptococcus Pyogenes Escapes from Autophagy. Cell Host Microbe 2013, 14, 604–606. [Google Scholar] [CrossRef] [Green Version]
- Baxt, L.A.; Goldberg, M.B. Host and Bacterial Proteins That Repress Recruitment of LC3 to Shigella Early during Infection. PLoS ONE 2014, 9, e94653. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Lenzen, G.; Page, A.-L.; Legrain, P.; Sansonetti, P.J.; Parsot, C. The Shigella Flexneri Effector OspG Interferes with Innate Immune Responses by Targeting Ubiquitin-Conjugating Enzymes. Proc. Natl. Acad. Sci. USA 2005, 102, 14046–14051. [Google Scholar] [CrossRef] [Green Version]
- Mostowy, S.; Sancho-Shimizu, V.; Hamon, M.A.; Simeone, R.; Brosch, R.; Johansen, T.; Cossart, P. P62 and NDP52 Proteins Target Intracytosolic Shigella and Listeria to Different Autophagy Pathways. J. Biol. Chem. 2011, 286, 26987–26995. [Google Scholar] [CrossRef] [Green Version]
- Okuda, J.; Toyotome, T.; Kataoka, N.; Ohno, M.; Abe, H.; Shimura, Y.; Seyedarabi, A.; Pickersgill, R.; Sasakawa, C. Shigella Effector IpaH9.8 Binds to a Splicing Factor U2AF(35) to Modulate Host Immune Responses. Biochem. Biophys. Res. Commun. 2005, 333, 531–539. [Google Scholar] [CrossRef]
- Catic, A.; Misaghi, S.; Korbel, G.A.; Ploegh, H.L. ElaD, a Deubiquitinating Protease Expressed by E. Coli. PLoS ONE 2007, 2, e381. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Ogawa, M.; Hain, T.; Yoshida, M.; Fukumatsu, M.; Kim, M.; Mimuro, H.; Nakagawa, I.; Yanagawa, T.; Ishii, T.; et al. Listeria Monocytogenes ActA-Mediated Escape from Autophagic Recognition. Nat. Cell Biol. 2009, 11, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Dortet, L.; Mostowy, S.; Louaka, A.S.; Gouin, E.; Nahori, M.-A.; Wiemer, E.A.C.; Dussurget, O.; Cossart, P. Recruitment of the Major Vault Protein by InlK: A Listeria Monocytogenes Strategy to Avoid Autophagy. PLoS Pathog. 2011, 7, e1002168. [Google Scholar] [CrossRef]
- Le Negrate, G.; Krieg, A.; Faustin, B.; Loeffler, M.; Godzik, A.; Krajewski, S.; Reed, J.C. ChlaDub1 of Chlamydia Trachomatis Suppresses NF-KappaB Activation and Inhibits IkappaBalpha Ubiquitination and Degradation. Cell. Microbiol. 2008, 10, 1879–1892. [Google Scholar] [CrossRef] [PubMed]
- Furtado, A.R.; Essid, M.; Perrinet, S.; Balañá, M.E.; Yoder, N.; Dehoux, P.; Subtil, A. The Chlamydial OTU Domain-Containing Protein ChlaOTU Is an Early Type III Secretion Effector Targeting Ubiquitin and NDP52. Cell. Microbiol. 2013, 15, 2064–2079. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Aravind, L.; Koonin, E.V. A Novel Superfamily of Predicted Cysteine Proteases from Eukaryotes, Viruses and Chlamydia Pneumoniae. Trends Biochem. Sci. 2000, 25, 50–52. [Google Scholar] [CrossRef]
- Laskowski-Arce, M.A.; Orth, K. The Elusive Activity of the Yersinia Protein Kinase A Kinase Domain Is Revealed. Trends Microbiol. 2007, 15, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Bassères, E.; Coppotelli, G.; Pfirrmann, T.; Andersen, J.B.; Masucci, M.; Frisan, T. The Ubiquitin C-Terminal Hydrolase UCH-L1 Promotes Bacterial Invasion by Altering the Dynamics of the Actin Cytoskeleton. Cell. Microbiol. 2010, 12, 1622–1633. [Google Scholar] [CrossRef]
- Kummari, E.; Alugubelly, N.; Hsu, C.-Y.; Dong, B.; Nanduri, B.; Edelmann, M.J. Activity-Based Proteomic Profiling of Deubiquitinating Enzymes in Salmonella-Infected Macrophages Leads to Identification of Putative Function of UCH-L5 in Inflammasome Regulation. PLoS ONE 2015, 10, e0135531. [Google Scholar] [CrossRef]
- Coombs, N.; Sompallae, R.; Olbermann, P.; Gastaldello, S.; Göppel, D.; Masucci, M.G.; Josenhans, C. Helicobacter Pylori Affects the Cellular Deubiquitinase USP7 and Ubiquitin-Regulated Components TRAF6 and the Tumour Suppressor P53. Int. J. Med. Microbiol. 2011, 301, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.; Oyston, P.C.F.; Green, M.; Titball, R.W. Tularemia. Clin. Microbiol. Rev. 2002, 15, 631–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechous, R.D.; McCarthy, T.R.; Zahrt, T.C. Working toward the Future: Insights into Francisella Tularensis Pathogenesis and Vaccine Development. Microbiol. Mol. Biol. Rev. 2009, 73, 684–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putzova, D.; Panda, S.; Härtlova, A.; Stulík, J.; Gekara, N.O. Subversion of Innate Immune Responses by Francisella Involves the Disruption of TRAF3 and TRAF6 Signalling Complexes. Cell. Microbiol. 2017, 19, e12769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshraghi, A.; Kim, J.; Walls, A.C.; Ledvina, H.E.; Miller, C.N.; Ramsey, K.M.; Whitney, J.C.; Radey, M.C.; Peterson, S.B.; Ruhland, B.R.; et al. Secreted Effectors Encoded within and Outside of the Francisella Pathogenicity Island Promote Intramacrophage Growth. Cell Host Microbe 2016, 20, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, Y.; Liang, H.; Xu, T.; Kong, Y.; Huang, M.; Xiao, J.; Chen, X.; Xia, H.; Wu, Y.; et al. HECTD3 Mediates TRAF3 Polyubiquitination and Type I Interferon Induction during Bacterial Infection. J. Clin. Investig. 2018, 128, 4148–4162. [Google Scholar] [CrossRef] [Green Version]
- Akimana, C.; Al-Khodor, S.; Abu Kwaik, Y. Host Factors Required for Modulation of Phagosome Biogenesis and Proliferation of Francisella Tularensis within the Cytosol. PLoS ONE 2010, 5, e11025. [Google Scholar] [CrossRef]
- Guo, Y.; Li, L.; Xu, T.; Guo, X.; Wang, C.; Li, Y.; Yang, Y.; Yang, D.; Sun, B.; Zhao, X.; et al. HUWE1 Mediates Inflammasome Activation and Promotes Host Defense against Bacterial Infection. J. Clin. Investig. 2020, 130, 6301–6316. [Google Scholar] [CrossRef] [PubMed]
- Woolard, M.D.; Wilson, J.E.; Hensley, L.L.; Jania, L.A.; Kawula, T.H.; Drake, J.R.; Frelinger, J.A. Francisella Tularensis-Infected Macrophages Release Prostaglandin E2 That Blocks T Cell Proliferation and Promotes a Th2-like Response. J. Immunol. 2007, 178, 2065–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.E.; Katkere, B.; Drake, J.R. Francisella Tularensis Induces Ubiquitin-Dependent Major Histocompatibility Complex Class II Degradation in Activated Macrophages. Infect. Immun. 2009, 77, 4953–4965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, D.; Wilson, J.E.; Weih, K.A.; Ishido, S.; Harton, J.A.; Roche, P.A.; Drake, J.R. Francisella Tularensis Elicits IL-10 via a PGE2-Inducible Factor, to Drive Macrophage MARCH1 Expression and Class II Down-Regulation. PLoS ONE 2012, 7, e37330. [Google Scholar] [CrossRef]
- Casabona, M.G.; Buchanan, G.; Zoltner, M.; Harkins, C.P.; Holden, M.T.G.; Palmer, T. Functional Analysis of the EsaB Component of the Staphylococcus aureus Type VII Secretion System. Microbiology 2017, 163, 1851–1863. [Google Scholar] [CrossRef] [Green Version]
- Warne, B.; Harkins, C.P.; Harris, S.R.; Vatsiou, A.; Stanley-Wall, N.; Parkhill, J.; Peacock, S.J.; Palmer, T.; Holden, M.T.G. The Ess/Type VII Secretion System of Staphylococcus aureus Shows Unexpected Genetic Diversity. BMC Genomics 2016, 17, 222. [Google Scholar] [CrossRef] [Green Version]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2–Keap1 Antioxidant Response by the Ubiquitin Proteasome System: An Insight into Cullin-Ring Ubiquitin Ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear Heme Oxygenase-1 (HO-1) Modulates Subcellular Distribution and Activation of Nrf2, Impacting Metabolic and Anti-Oxidant Defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.W.; Hall, S.; Perrella, M.A. Role of Heme Oxygenase-1 in Microbial Host Defense. Cell. Microbiol. 2009, 11, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharn, C.R.; Collins, A.C.; Nair, V.R.; Stamm, C.E.; Marciano, D.K.; Graviss, E.A.; Shiloh, M.U. Heme Oxygenase-1 Regulates Inflammation and Mycobacterial Survival in Human Macrophages during M. Tuberculosis Infection. J. Immunol. 2016, 196, 4641–4649. [Google Scholar] [CrossRef] [Green Version]
- Lee, C. Therapeutic Modulation of Virus-Induced Oxidative Stress via the Nrf2-Dependent Antioxidative Pathway. Oxid. Med. Cell. Longev. 2018, 2018, 6208067. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy Revisited: A Conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and Human Diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Murrow, L.; Debnath, J. Autophagy as a Stress-Response and Quality-Control Mechanism: Implications for Cell Injury and Human Disease. Annu. Rev. Pathol. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J. Autophagy: From Phenomenology to Molecular Understanding in Less than a Decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. A Brief History of Autophagy from Cell Biology to Physiology and Disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I. Ubiquitin Signaling and Autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate MTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Radoshevich, L.; Cossart, P. Listeria Monocytogenes: Towards a Complete Picture of Its Physiology and Pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.; Ge, L.; Huang, Q.; Chen, C.; Kianian, S.; Roberts, M.F.; Schekman, R.; Portnoy, D.A. Avoidance of Autophagy Mediated by PlcA or ActA Is Required for Listeria Monocytogenes Growth in Macrophages. Infect. Immun. 2015, 83, 2175–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, M.; Yoshimori, T.; Suzuki, T.; Sagara, H.; Mizushima, N.; Sasakawa, C. Escape of Intracellular Shigella from Autophagy. Science 2005, 307, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Zhu, Y.; Lu, Q.; Hu, L.; Zheng, Y.; Shao, F. Structurally Distinct Bacterial TBC-like GAPs Link Arf GTPase to Rab1 Inactivation to Counteract Host Defenses. Cell 2012, 150, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. P62/SQSTM1 Forms Protein Aggregates Degraded by Autophagy and Has a Protective Effect on Huntingtin-Induced Cell Death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. P62 Links the Autophagy Pathway and the Ubiqutin–Proteasome System upon Ubiquitinated Protein Degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.-Z.; Jiang, A.-J.; Mao, A.-W.; Feng, Y.; Wang, W.; Li, J.; Zhang, X.; Xing, K.; Peng, X. The Salmonella Effectors SseF and SseG Inhibit Rab1A-Mediated Autophagy to Facilitate Intracellular Bacterial Survival and Replication. J. Biol. Chem. 2018, 293, 9662–9673. [Google Scholar] [CrossRef] [Green Version]
- Choy, A.; Dancourt, J.; Mugo, B.; O’Connor, T.J.; Isberg, R.R.; Melia, T.J.; Roy, C.R. The Legionella Effector RavZ Inhibits Host Autophagy through Irreversible Atg8 Deconjugation. Science 2012, 338, 1072–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A Ubiquitin-like System Mediates Protein Lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Horenkamp, F.A.; Kauffman, K.J.; Kohler, L.J.; Sherwood, R.K.; Krueger, K.P.; Shteyn, V.; Roy, C.R.; Melia, T.J.; Reinisch, K.M. The Legionella Anti-Autophagy Effector RavZ Targets the Autophagosome via PI3P- and Curvature-Sensing Motifs. Dev. Cell 2015, 34, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Pantoom, S.; Wu, Y.-W. Elucidation of the Anti-Autophagy Mechanism of the Legionella Effector RavZ Using Semisynthetic LC3 Proteins. eLife 2017, 6, e23905. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Fraunholz, M. Staphylococcus aureus Host Cell Invasion and Post-Invasion Events. Int. J. Med. Microbiol. IJMM 2010, 300, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Neumann, Y.; Bruns, S.A.; Rohde, M.; Prajsnar, T.K.; Foster, S.J.; Schmitz, I. Intracellular Staphylococcus aureus Eludes Selective Autophagy by Activating a Host Cell Kinase. Autophagy 2016, 12, 2069–2084. [Google Scholar] [CrossRef] [Green Version]
- Chong, A.; Wehrly, T.D.; Child, R.; Hansen, B.; Hwang, S.; Virgin, H.W.; Celli, J. Cytosolic Clearance of Replication-Deficient Mutants Reveals Francisella Tularensis Interactions with the Autophagic Pathway. Autophagy 2012, 8, 1342–1356. [Google Scholar] [CrossRef] [Green Version]
- Case, E.D.R.; Chong, A.; Wehrly, T.D.; Hansen, B.; Child, R.; Hwang, S.; Virgin, H.W.; Celli, J. The Francisella O-Antigen Mediates Survival in the Macrophage Cytosol via Autophagy Avoidance. Cell. Microbiol. 2014, 16, 862–877. [Google Scholar] [CrossRef] [Green Version]
- Chai, Q.; Wang, X.; Qiang, L.; Zhang, Y.; Ge, P.; Lu, Z.; Zhong, Y.; Li, B.; Wang, J.; Zhang, L.; et al. A Mycobacterium Tuberculosis Surface Protein Recruits Ubiquitin to Trigger Host Xenophagy. Nat. Commun. 2019, 10, 1973. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, J.; Li, J.; Yang, H.; Tang, T.; Liang, H.; Zuo, M.; Wang, J.; Liu, H.; Liu, F.; et al. Host-Mediated Ubiquitination of a Mycobacterial Protein Suppresses Immunity. Nature 2020, 577, 682–688. [Google Scholar] [CrossRef]
- Wang, J.; Li, B.-X.; Ge, P.-P.; Li, J.; Wang, Q.; Gao, G.F.; Qiu, X.-B.; Liu, C.H. Mycobacterium Tuberculosis Suppresses Innate Immunity by Coopting the Host Ubiquitin System. Nat. Immunol. 2015, 16, 237–245. [Google Scholar] [CrossRef]
- Cornelis, G.R. The Yersinia Deadly Kiss. J. Bacteriol. 1998, 180, 5495–5504. [Google Scholar] [CrossRef] [Green Version]
- Orth, K. Function of the Yersinia Effector YopJ. Curr. Opin. Microbiol. 2002, 5, 38–43. [Google Scholar] [CrossRef]
- Lostroh, C.P.; Lee, C.A. The Salmonella Pathogenicity Island-1 Type III Secretion System. Microbes Infect. 2001, 3, 1281–1291. [Google Scholar] [CrossRef]
- Oh, Y.K.; Alpuche-Aranda, C.; Berthiaume, E.; Jinks, T.; Miller, S.I.; Swanson, J.A. Rapid and Complete Fusion of Macrophage Lysosomes with Phagosomes Containing Salmonella Typhimurium. Infect. Immun. 1996, 64, 3877–3883. [Google Scholar] [CrossRef] [Green Version]
- Rytkönen, A.; Poh, J.; Garmendia, J.; Boyle, C.; Thompson, A.; Liu, M.; Freemont, P.; Hinton, J.C.D.; Holden, D.W. SseL, a Salmonella Deubiquitinase Required for Macrophage Killing and Virulence. Proc. Natl. Acad. Sci. USA 2007, 104, 3502–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, A.P.; Petrof, E.O.; Kuppireddi, S.; Zhao, Y.; Xia, Y.; Claud, E.C.; Sun, J. Salmonella Type III Effector AvrA Stabilizes Cell Tight Junctions to Inhibit Inflammation in Intestinal Epithelial Cells. PLoS ONE 2008, 3, e2369. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Wu, H.; Wentworth, C.; Luo, L.; Collier-Hyams, L.; Neish, A.S. Salmonella AvrA Coordinates Suppression of Host Immune and Apoptotic Defenses via JNK Pathway Blockade. Cell Host Microbe 2008, 3, 233–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, S.; Wang, Y.; Xue, Y.; Wang, H.; Cai, Y.; Zhang, J.; Barrow, P.; Pan, Z.; Jiao, X. The SseL Protein Inhibits the Intracellular NF-ΚB Pathway to Enhance the Virulence of Salmonella Pullorum in a Chicken Model. Microb. Pathog. 2019, 129, 1–6. [Google Scholar] [CrossRef]
- Kolodziejek, A.M.; Altura, M.A.; Fan, J.; Petersen, E.M.; Cook, M.; Brzovic, P.S.; Miller, S.I. Salmonella Translocated Effectors Recruit OSBP1 to the Phagosome to Promote Vacuolar Membrane Integrity. Cell Rep. 2019, 27, 2147–2156.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanns, T.; Hofmann, K. Bacterial DUBs: Deubiquitination beyond the Seven Classes. Biochem. Soc. Trans. 2019, 47, 1857–1866. [Google Scholar] [CrossRef] [Green Version]
- Vogel, J.P.; Andrews, H.L.; Wong, S.K.; Isberg, R.R. Conjugative Transfer by the Virulence System of Legionella Pneumophila. Science 1998, 279, 873–876. [Google Scholar] [CrossRef]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.-Q. Ubiquitination Independent of E1 and E2 Enzymes by Bacterial Effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Mu, Y.; Xie, Y.; Zhang, Y.; Han, Y.; Zhou, Y.; Wang, W.; Liu, Z.; Wu, M.; Wang, H.; et al. Structural Basis of Ubiquitin Modification by the Legionella Effector SdeA. Nature 2018, 557, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Puvar, K.; Iyer, S.; Sheedlo, M.J.; Das, C. Chapter Fifteen—Purification and functional characterization of the DUB domain of SdeA. In Methods in Enzymology; Hochstrasser, M., Ed.; Ubiquitin and Ubiquitin-Like Protein Modifiers; Academic Press: Cambridge, MA, USA, 2019; Volume 618, pp. 343–355. [Google Scholar]
- Pike, C.M.; Boyer-Andersen, R.; Kinch, L.N.; Caplan, J.L.; Neunuebel, M.R. The Legionella Effector RavD Binds Phosphatidylinositol-3-Phosphate and Helps Suppress Endolysosomal Maturation of the Legionella-Containing Vacuole. J. Biol. Chem. 2019, 294, 6405–6415. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.N.; Hilbi, H. Molecular Pathogenesis of Shigella Spp.: Controlling Host Cell Signaling, Invasion, and Death by Type III Secretion. Clin. Microbiol. Rev. 2008, 21, 134–156. [Google Scholar] [CrossRef] [Green Version]
- Ashida, H.; Toyotome, T.; Nagai, T.; Sasakawa, C. Shigella Chromosomal IpaH Proteins Are Secreted via the Type III Secretion System and Act as Effectors. Mol. Microbiol. 2007, 63, 680–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, F.; Iwai, K. LUBAC, a Novel Ubiquitin Ligase for Linear Ubiquitination, Is Crucial for Inflammation and Immune Responses. Microbes Infect. 2012, 14, 563–572. [Google Scholar] [CrossRef]
- Ashida, H.; Kim, M.; Schmidt-Supprian, M.; Ma, A.; Ogawa, M.; Sasakawa, C. A Bacterial E3 Ubiquitin Ligase IpaH9.8 Targets NEMO/IKKgamma to Dampen the Host NF-KappaB-Mediated Inflammatory Response. Nat. Cell Biol. 2010, 12, 66–73. [Google Scholar] [CrossRef] [Green Version]
- De Jong, M.F.; Liu, Z.; Chen, D.; Alto, N.M. Shigella Flexneri Suppresses NF-KB Activation by Inhibiting Linear Ubiquitin Chain Ligation. Nat. Microbiol. 2016, 1, 16084. [Google Scholar] [CrossRef] [Green Version]
- Noad, J.; von der Malsburg, A.; Pathe, C.; Michel, M.A.; Komander, D.; Randow, F. LUBAC-Synthesized Linear Ubiquitin Chains Restrict Cytosol-Invading Bacteria by Activating Autophagy and NF-ΚB. Nat. Microbiol. 2017, 2, 17063. [Google Scholar] [CrossRef] [Green Version]
- Misaghi, S.; Balsara, Z.R.; Catic, A.; Spooner, E.; Ploegh, H.L.; Starnbach, M.N. Chlamydia Trachomatis-Derived Deubiquitinating Enzymes in Mammalian Cells during Infection. Mol. Microbiol. 2006, 61, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Pruneda, J.N.; Bastidas, R.J.; Bertsoulaki, E.; Swatek, K.N.; Santhanam, B.; Clague, M.J.; Valdivia, R.H.; Urbé, S.; Komander, D. A Chlamydia Effector Combining Deubiquitination and Acetylation Activities Induces Golgi Fragmentation. Nat. Microbiol. 2018, 3, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Harrison, K.S.; Ramirez, Y.; Auer, D.; Chowdhury, S.R.; Prusty, B.K.; Sauer, F.; Dimond, Z.; Kisker, C.; Hefty, P.S.; et al. Chlamydia Trachomatis-Containing Vacuole Serves as Deubiquitination Platform to Stabilize Mcl-1 and to Interfere with Host Defense. eLife 2017, 6, e21465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (VelcadeTM) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the First Proteasome Inhibitor Anticancer Drug: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Groen, K.; van de Donk, N.; Stege, C.; Zweegman, S.; Nijhof, I.S. Carfilzomib for Relapsed and Refractory Multiple Myeloma. Cancer Manag. Res. 2019, 11, 2663–2675. [Google Scholar] [CrossRef] [Green Version]
- Nawrocki, S.T.; Griffin, P.; Kelly, K.R.; Carew, J.S. MLN4924: A Novel First-in-Class Inhibitor of NEDD8-Activating Enzyme for Cancer Therapy. Expert Opin. Investig. Drugs 2012, 21, 1563–1573. [Google Scholar] [CrossRef]
- Millennium Pharmaceuticals, Inc. MLN4924 for the Treatment of Acute Myelogenous Leukemia, Myelodysplastic Syndrome, and Acute Lymphoblastic Leukemia; Millennium Pharmaceuticals Inc.: Cambridge, MA, USA, 2013. [Google Scholar]
- Zhuang, J.; Shirazi, F.; Singh, R.K.; Kuiatse, I.; Wang, H.; Lee, H.C.; Berkova, Z.; Berger, A.; Hyer, M.; Chattopadhyay, N.; et al. Ubiquitin-Activating Enzyme Inhibition Induces an Unfolded Protein Response and Overcomes Drug Resistance in Myeloma. Blood 2019, 133, 1572–1584. [Google Scholar] [CrossRef] [Green Version]
- Charbonneau, M.-E.; Gonzalez-Hernandez, M.J.; Showalter, H.D.; Donato, N.J.; Wobus, C.E.; O’Riordan, M.X.D. Small Molecule Deubiquitinase Inhibitors Promote Macrophage Anti-Infective Capacity. PLoS ONE 2014, 9, e104096. [Google Scholar] [CrossRef]
- Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Talpaz, M.; Donato, N.J. Deubiquitinase Inhibition by Small-Molecule WP1130 Triggers Aggresome Formation and Tumor Cell Apoptosis. Cancer Res. 2010, 70, 9265–9276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, H.A. Deubiquitination in Virus Infection. Virology 2007, 362, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Longhitano, L.; Tibullo, D.; Giallongo, C.; Lazzarino, G.; Tartaglia, N.; Galimberti, S.; Li Volti, G.; Palumbo, G.A.; Liso, A. Proteasome Inhibitors as a Possible Therapy for SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 3622. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effector Protein | Bacteria | Role | Function | References |
|---|---|---|---|---|
| YpkA | Yersinia pseudotuberculosis, Y. enterocolitica | Protein kinase | Increasing of bacterial uptake | [57] |
| YopJ | Yersinia pseudotuberculosis | DUB, acetyltransferase | Suppression of MAPK and NF-κB pathway | [58,59] |
| SopA | Salmonella Typhimurium | E3 ligase | Unknown | [60] |
| SseL | Salmonella Typhimurium | DUB | Suppression of NF-κB pathway | [61] |
| Inhibition of autophagy | [62] | |||
| AvrA | Salmonella Typhimurium | DUB, acetyltransferase | Suppression of NF-κB pathway and stabilization of β-catenin | [63] |
| NleL | Escherichia coli | E3 ligase | Suppression of the inflammatory response | [64] |
| ElaD | Escherichia coli | DUB | Suppression of NF-κB pathway | [54] |
| SidE | Legionella pneumophila | E3 ligase | Regulation of phosphoribosyl ubiquitination, cytotoxicity | [65,66] |
| DupA, B | Legionella pneumophila | DUB | ||
| SdeA | Legionella pneumophila | DUB | Inhibition of autophagy | [67] |
| LotA | Legionella pneumophila | DUB | Suppression of phagosome maturation | [68] |
| RavD | Legionella pneumophila | DUB | Suppression of phagosome maturation and NF-κB pathway | [69] |
| RavZ | Legionella pneumophila | DUB | Inhibition of autophagy | [70] |
| SpeB | Streptococcus pyogenes | Cysteine protease | Inhibition of autophagy | [71] |
| IcsB | Shigella flexneri | Virulence factor | Inhibition of autophagy | [72] |
| OspG | Shigella flexneri | E2 enzyme | Suppression of NF-κB pathway | [73] |
| IpaH | Shigella flexneri | E3 ligase | Suppression of NF-κB pathway and autophagy | [74,75] |
| SchiCE | Shigella flexneri | DUB | Suppression of NF-κB pathway | [54,76] |
| ActA | Listeria monocytogenes | Virulence factor | Increasing of intracellular motility to avoiding autophagy | [77] |
| InlK | Listeria monocytogenes | Virulence factor | Inhibition of autophagy | [78] |
| ChlaDUB1 | Chlamydia trachomatis | DUB, acetyltransferase | Suppression of NF-κB pathway | [79] |
| ChlaDUB2 | Chlamydia trachomatis | DUB | Suppression of NF-κB pathway | |
| ChlaOTU | Chlamydia pneumonia | DUB | Unknown | [80,81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vozandychova, V.; Stojkova, P.; Hercik, K.; Rehulka, P.; Stulik, J. The Ubiquitination System within Bacterial Host–Pathogen Interactions. Microorganisms 2021, 9, 638. https://doi.org/10.3390/microorganisms9030638
Vozandychova V, Stojkova P, Hercik K, Rehulka P, Stulik J. The Ubiquitination System within Bacterial Host–Pathogen Interactions. Microorganisms. 2021; 9(3):638. https://doi.org/10.3390/microorganisms9030638
Chicago/Turabian StyleVozandychova, Vera, Pavla Stojkova, Kamil Hercik, Pavel Rehulka, and Jiri Stulik. 2021. "The Ubiquitination System within Bacterial Host–Pathogen Interactions" Microorganisms 9, no. 3: 638. https://doi.org/10.3390/microorganisms9030638
APA StyleVozandychova, V., Stojkova, P., Hercik, K., Rehulka, P., & Stulik, J. (2021). The Ubiquitination System within Bacterial Host–Pathogen Interactions. Microorganisms, 9(3), 638. https://doi.org/10.3390/microorganisms9030638