
Metagenomic Analysis of the Respiratory Microbiome of a Broiler Flock from Hatching to Processing

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Nucleic Acid Extraction and Sequencing
2.3. 16S rRNA Amplicon Sequencing and Analysis
2.4. Eukaryotic Virus, Bacteriophage and Fungal Analysis
3. Results
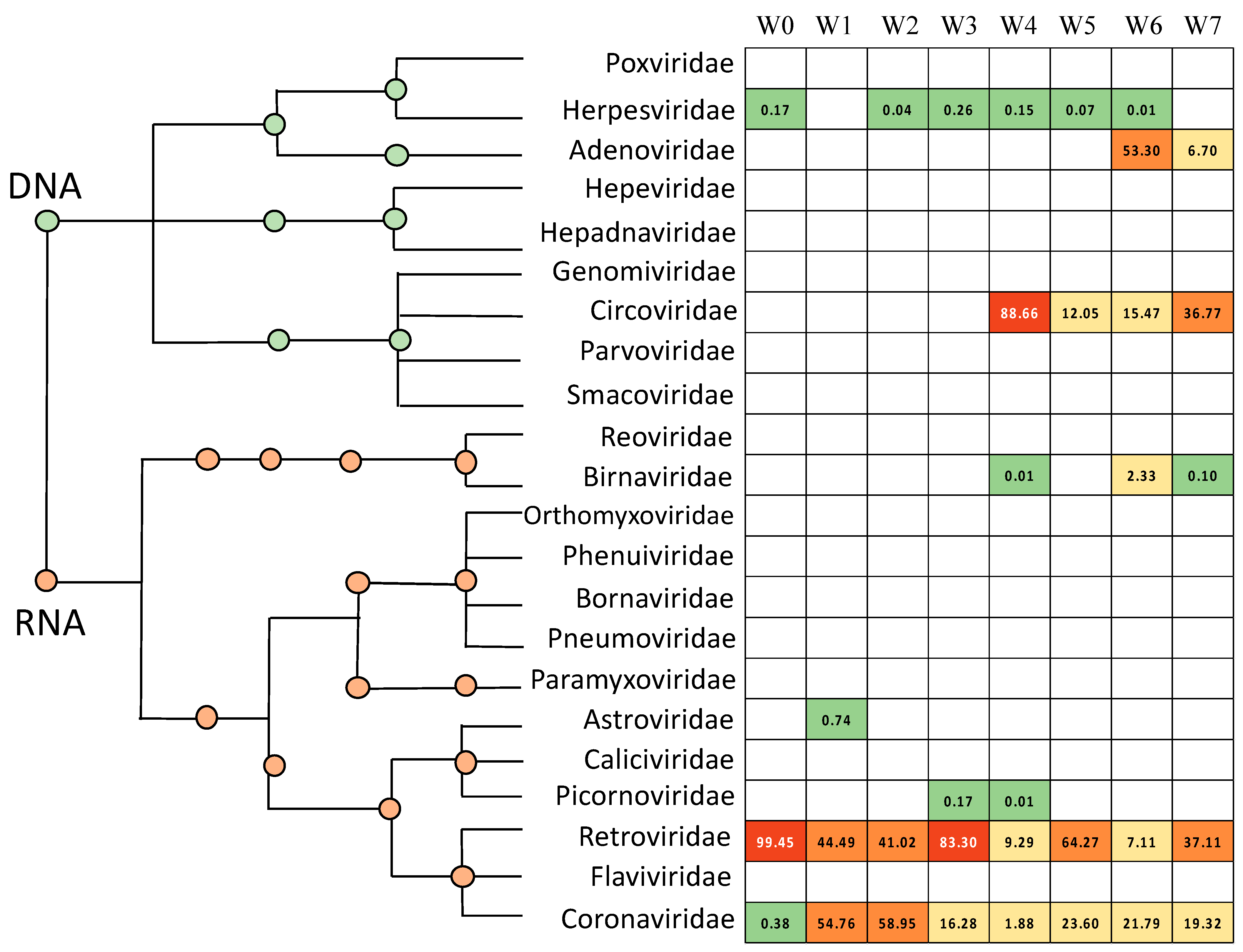
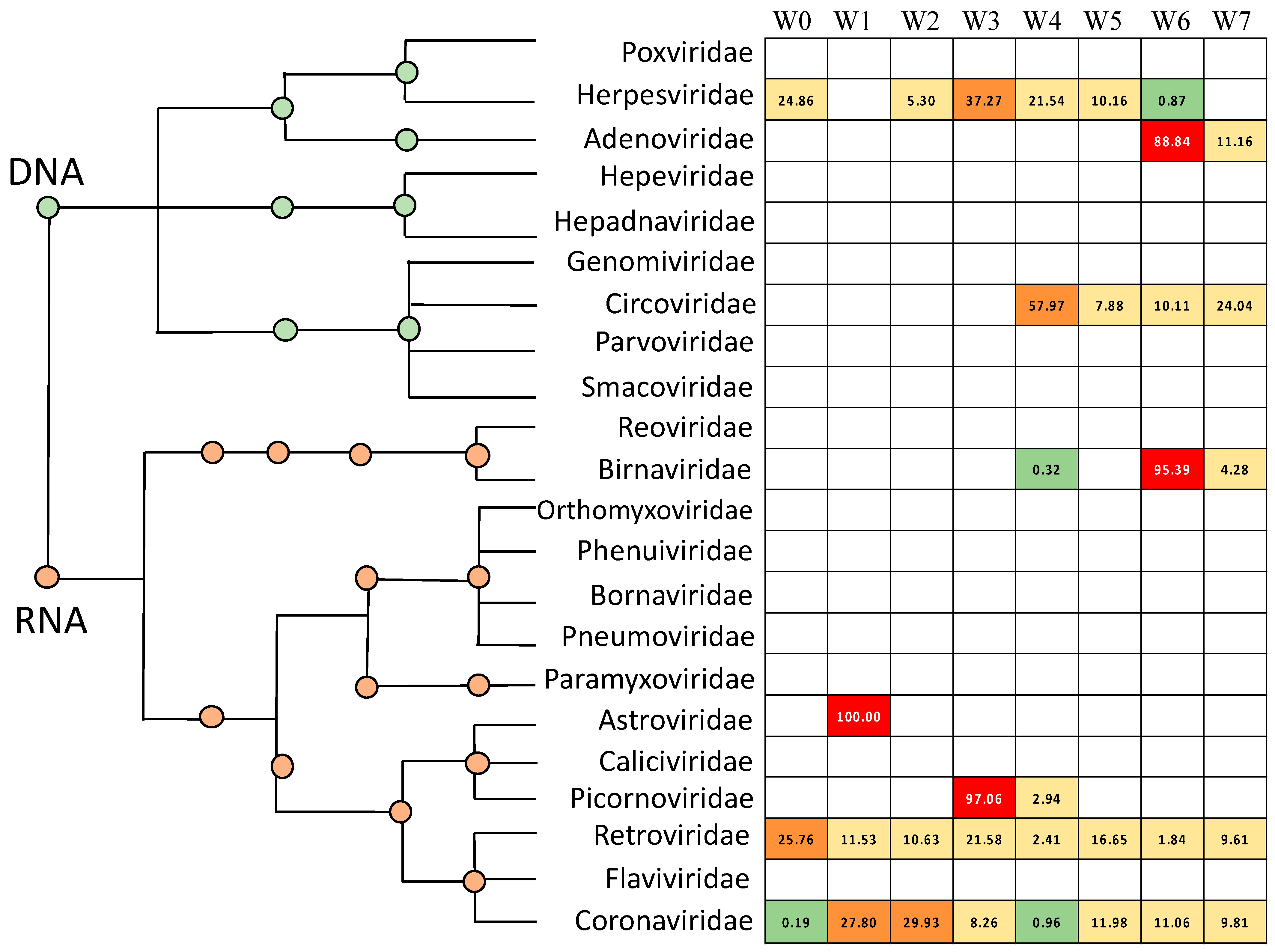
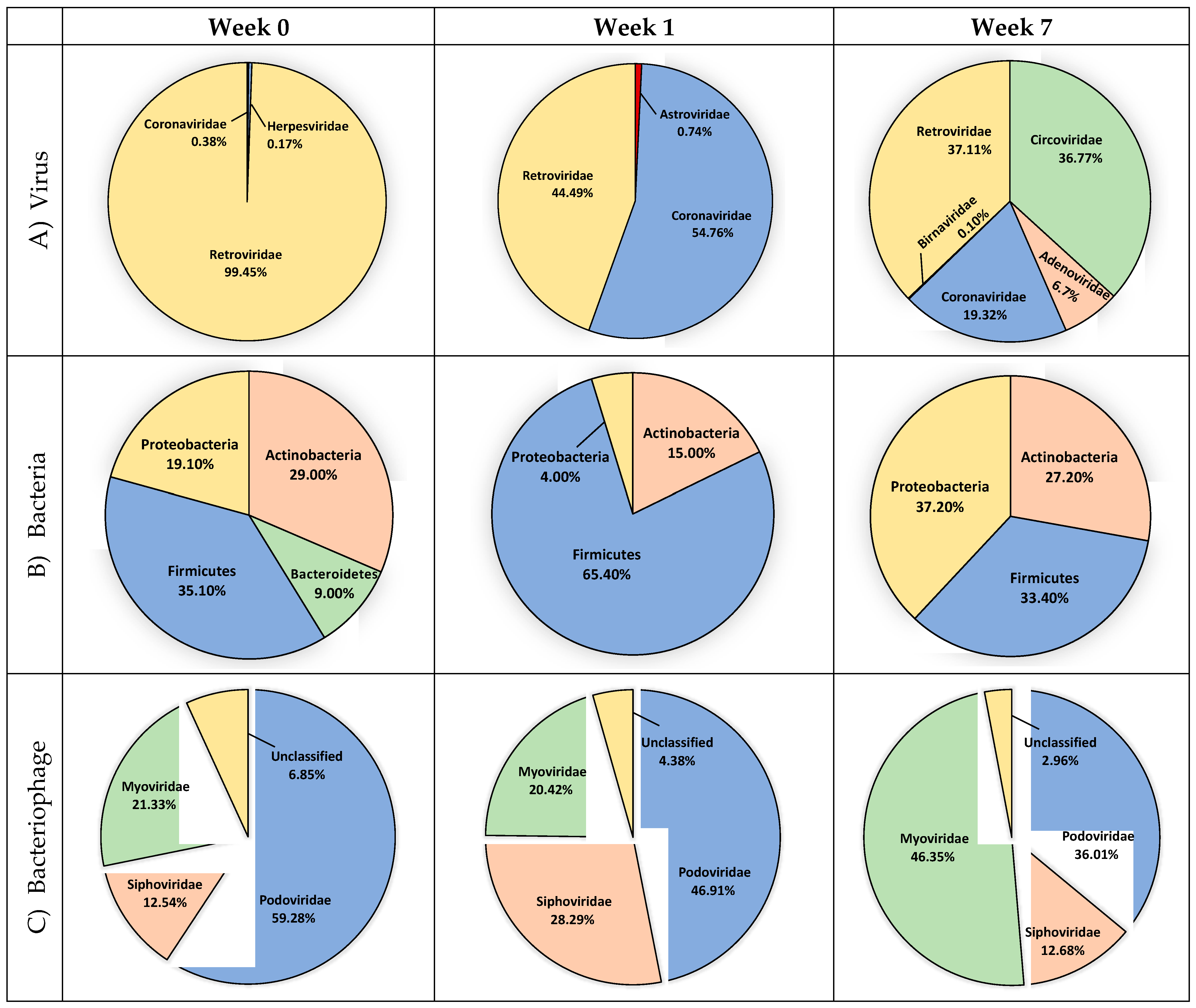
3.1. Avian Respiratory Eukaryotic Viral Diversity
3.2. Bacterial Diversity
3.3. Bacteriophage Diversity
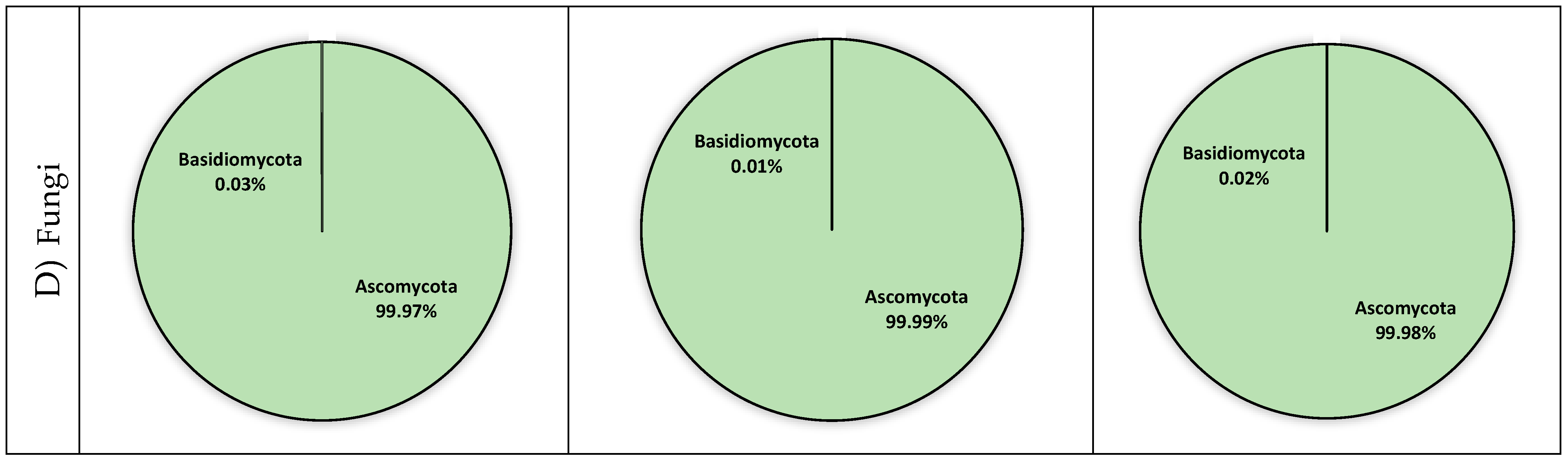
3.4. Fungal Diversity
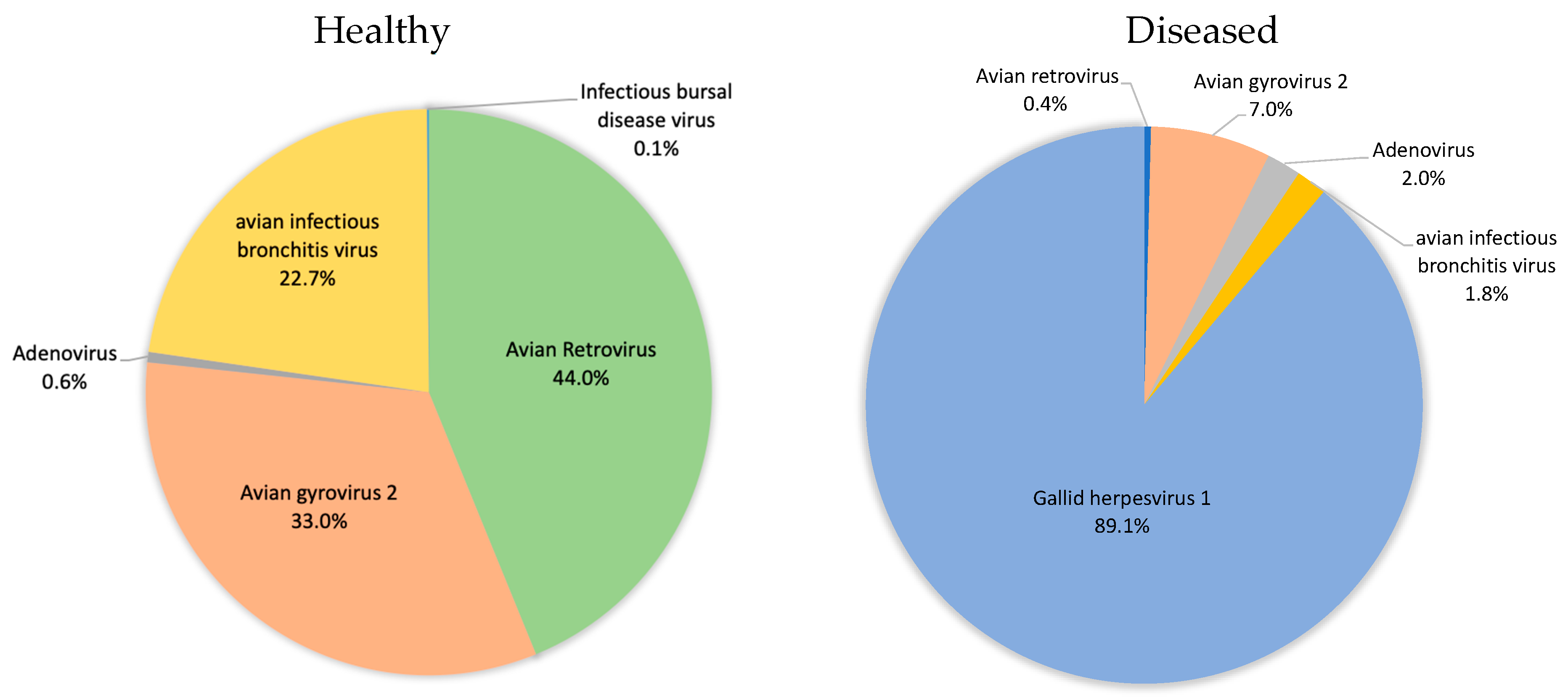
3.5. Poultry with Respiratory Disease Exhibit a Dysbiosis of the Respiratory Virome
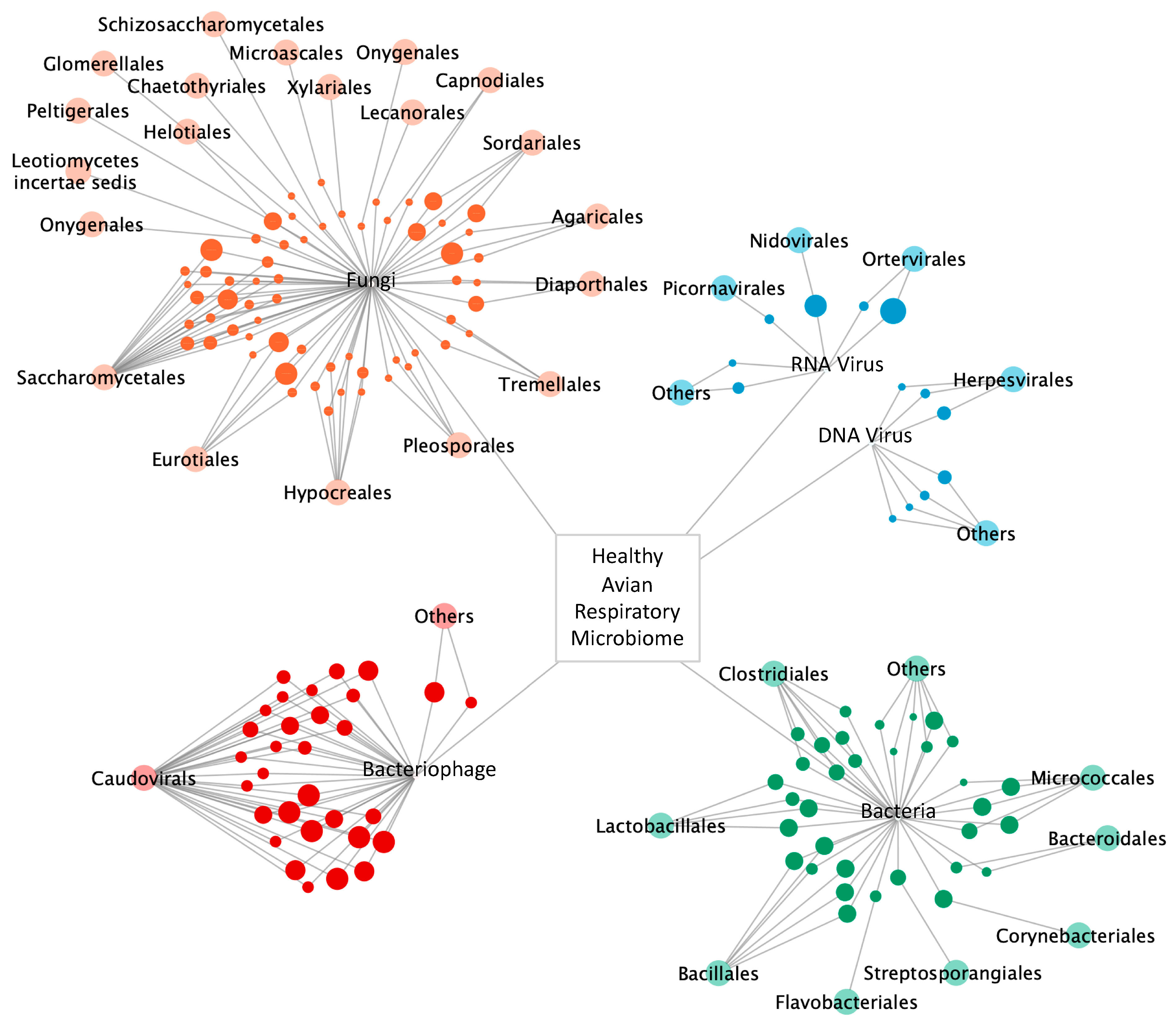
3.6. The Avian Microbiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Micah, H.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Shafquat, A.; Joice, R.; Simmons, S.L.; Huttenhower, C. Functional and phylogenetic assembly of microbial communities in the human microbiome. Trends Microbiol. 2014, 22, 261–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, E.S.W.D.W.; Cathelyn, J.S.; Brett-McClellan, K.A.; Engle, M.; Diamond, M.S.; Miller, V.L.; Virgin, H.W. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 2007, 447, 326–329. [Google Scholar] [CrossRef]
- Yager, E.J.; Szaba, F.M.; Kummer, L.W.; Lanzer, K.G.; Burkum, C.E.; Smiley, S.T.; Blackman, M.A. Gamma-Herpesvirus-induced protection against bacterial infection is transient. Viral Immunol. 2009, 22, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Grivel, J.; Ito, Y.; Fagà, G. Suppression of CCR5- but not CXCR4-tropic HIV-1 in lymphoid tissue by human herpesvirus 6. Nat. Med. 2001, 7, 1232–1235. [Google Scholar] [CrossRef]
- Gross, W.B. Factors affecting the development of respiratory disease complex in chickens. Avian Dis. 1990, 34, 607–610. [Google Scholar] [CrossRef]
- Hatoum, R.; Labrie, S.; Fliss, I. Antimicrobial and probiotic properties of yeasts: From fundamental to novel applications. Front. Microbiol. 2012, 3, 421. [Google Scholar] [CrossRef] [Green Version]
- McFarland, L.V. Systematic review and meta-analysis of Saccharomyces boulardii in adult patients. World J. Gastroenterol. 2010, 16, 2202–2222. [Google Scholar] [CrossRef]
- Bradley, G.L.; Savage, T.F.T.K.I. The effects of supplementing diets with Saccharomyces cerevisiae var. boulardii on male poult performance and ileal morphology. Poult. Sci. 1994, 73, 1766–1770. [Google Scholar]
- Bond, S.; Timsit, E.; Workentine, M.; Alexander, T.; Léguillette, R. Upper and lower respiratory tract microbiota in horses: Bacterial communities associated with health and mild asthma (inflammatory airway disease) and effects of dexamethasone. BMC Microbiol. 2017, 17, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boeck, C.; Kalmar, I.; Dumont, A.; Vanrompay, D. Longitudinal monitoring for respiratory pathogens in broiler chickens reveals co-infection of Chlamydia psittaci and Ornithobacterium rhinotracheale. J. Med. Microbio. 2015, 64, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaeta, N.; Lima, S.; Teixeira, A.; Ganda, E.; Oikonomou, G.; Gregory, L.; Bichalho, R. Deciphering upper respiratory tract microbiota complexity in healthy calves and calves that develop respiratory disease using shotgun metagenomics. J. Dairy Sci. 2017, 100, 1445–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glendinning, L.; McLachlan, G.; Vervelde, L. Age-related differences in the respiratory microbiota of chickens. PLoS ONE 2017, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.J.; Youmans, B.P.; Noll, S.; Cardona, C.; Evans, N.P.; Karnezos, P.; Ngunjiri, J.M.; Abundo, M.C.; Lee, C.-W. A consistent and predictable commercial broiler chicken bacterial microbiota in antibiotic-free production displays strong correlations with performance. Appl. Environ. Micro. 2018, 84, e00362-18. [Google Scholar] [CrossRef] [Green Version]
- Shabbir, M.; Malys, T.; Ivanov, Y.; Park, J.; Shabbir, M.; Rabbani, M.; Yaqub, T.; Harvill, E. Microbial communities present in the lower respiratory tract of clinically healthy birds in Pakistan. Poult. Sci. 2015, 94, 612–620. [Google Scholar] [CrossRef]
- Clarridge, J. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 2004, 17, 840–862. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2016, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.; Kubal, M.; Paczian, T.; Rodriguqz, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server- a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.; Costello, E.; Fierer, N.; Peña, A.; Goodrich, J.; Gordon, J.; et al. Qiime allows analysis of high-throughout community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.; Westcott, S.; Ryabin, T.; Hall, J.; Hartman, M.; Hollister, E.; Lesniewski, R.; Oakley, B.; Parks, D.; Robinson, C.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Caler, L.; Colombini-Hatch, S.; Glynn, S.; Srinivas, P. Research on the human virome: Where are we and what is next. Microbiome 2016, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the fecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel cell polyomavirus and two novel polyomaviruses are chronically shed from human skin. Cell Host Microbe. 2010, 7, 509–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohl, D.M.; Vangay, P.; Garbe, J.; MacLean, A.; Hauge, A.; Becker, A.; Gould, T.J.; Clayton, J.B.; Johnson, T.J.; Hunter, R.; et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 2016, l34, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.; Westcott, S.; Baxter, N.; Highlander, S.; Schloss, P. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Lemos, L.N.; Fulthorpe, R.R.; Triplett, E.W.; Roesch, L.F.W. Rethinking microbial diversity analysis in the high throughput sequencing era. J. Microbiol. Meth. 2011, 86, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, J.A.; Reynolds, J.F. Statistical Ecology: A Primer in Methods and Computing; John Wiley & Sons: New York, NY, USA, 2008; pp. 85–106. [Google Scholar]
- Asnicar, F.; Weingart, G.; Tickle, T.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2013, 13, 2498–2504. [Google Scholar] [CrossRef]
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix (Version 0.84). 2017. Available online: https://cran.r-project.org/web/packages/corrplot/corrplot.pdf (accessed on 30 March 2021).
- Mulholland, K.A.; Keeler, C.L. BiomeSeq: A tool for the characterization of animal microbiomes from metagenomic data. BioRxiv 2019. [Google Scholar] [CrossRef]
- Bellott, D.; Skaletsky, H.; Cho, T.; Bron, L.; Locke, D.; Chen, N.; Galkina, S.; Pyntikova, T.; Koutseva, N.; Graves, T.; et al. Avian W and mammalian Y chromosomes convergently retained dosage-sensitive regulators. Nat. Genet. 2017, 49, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, G.; Leggett, R.; Rowe, W.; Stubbs, S.; Wilkinson, M.; Ramirez-Gonzalez, R.; Mario Caccamo Bernal, W.; Heeney, J. Host subtraction, filtering and assembly validations for novel viral discovery using next generation sequencing data. PLoS ONE 2015, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herath, D.; Jayasundara, D.; Ackland, D.; Saeed, I.; Tang, S.; Halgamuge, S. Assessing species diversity using metavirome data: Methods and challenges. Comp. Struc. Biotechnol. J. 2017, 15, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Xie, C.; Kirkness, E.; Biggs, W.; Wong, E.; Turpaz, Y.; Bloom, K.; Delwart, E.; Nelson, K.; Venter, J.; et al. The blood DNA virome in 8000 humans. PLoS Pathog. 2017, 13, e1006292. [Google Scholar] [CrossRef] [Green Version]
- Revell, L. Phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Yang, S.; Gao, X.; Meng, J.; Zhang, A.; Zhou, Y.; Long, M.; Li, B.; Deng, W.; Jin, L.; Zhao, S.; et al. Metagenomics analysis of bacteria, fungi, bacteriophages, and helminths in the gut of giant pandas. Front. Micro. 2018, 9, 1717. [Google Scholar] [CrossRef] [Green Version]
- Day, J.M.; Ballard, L.L.; Duke, M.V.; Scheffler, B.E.; Zsak, L. Metagenomic analysis of the turkey gut RNA virus community. Virol. J. 2010, 7, 313. [Google Scholar] [CrossRef] [Green Version]
- Day, J.M.; Zsak, L. Recent progress in the characterization of avian enteric viruses. Avian Dis. 2013, 57, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Classification | Virus Family | Complete Genomes | |||
|---|---|---|---|---|---|---|
| Avian DNA Viral Database | Double/Single Stranded a | Enveloped d | Hepeviridae | 1 | ||
| Hepadnaviridae | 1 | |||||
| Single Stranded | Non- Enveloped | Genomoviridae | 3 | |||
| Parvoviridae | 7 | |||||
| Circoviridae | 10 | |||||
| Smacoviridae | 3 | |||||
| Double Stranded | Enveloped | Poxviridae | 3 | |||
| Herpesviridae | 6 | |||||
| Double Stranded | Non- Enveloped | Adenoviridae | 14 | |||
| Avian RNA Viral Database | Double Stranded | Segmented c | Non- Enveloped | Reoviridae | 5 | |
| Birnaviridae | 1 | |||||
| Single Stranded | Positive b | Non- Segmented | Enveloped | Retroviridae | 5 | |
| Flaviviridae | 3 | |||||
| Coronaviridae | 5 | |||||
| Single Stranded | Positive | Non- Segmented | Non- Enveloped | Astroviridae | 5 | |
| Caliciviridae | 1 | |||||
| Picornaviridae | 17 | |||||
| Single Stranded | Negative | Segmented | Enveloped | Orthomyxoviridae | 16 | |
| Phenuiviridae | 1 | |||||
| Bornaviridae | 3 | |||||
| Pneumoviridae | 1 | |||||
| Single Stranded | Negative | Non- Segmented | Enveloped | Paramyxoviridae | 14 | |
| Time | RNA Virus | DNA Virus | Bacteria | Phage | Fungi |
|---|---|---|---|---|---|
| Placement | 0.041 | 0.000 | 2.707 | 2.218 | 0.022 |
| Week 1 | 1.290 | 0.000 | 2.468 | 2.531 | 0.286 |
| Week 2 | 1.108 | 0.000 | 2.111 | 0.096 | |
| Week 3 | 0.722 | 0.000 | 2.251 | 2.922 | 0.165 |
| Week 4 | 0.738 | 0.013 | 2.499 | 2.756 | 0.151 |
| Week 5 | 0.910 | 0.035 | 1.925 | 2.294 | 0.026 |
| Week 6 | 1.480 | 0.534 | 2.134 | 0.013 | |
| Week 7 | 1.092 | 0.867 | 1.935 | 2.087 | 0.134 |
| Sample | Trimmed for Quality | Map to Host | Not Mapped to Host | Map to Virus DB |
|---|---|---|---|---|
| DNA | 23,547,613 | 21,003,097 | 2,544,516 | 32,478 |
| RNA | 8,680,175 | 8,217,793 | 462,382 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mulholland, K.A.; Robinson, M.G.; Keeler, S.J.; Johnson, T.J.; Weber, B.W.; Keeler, C.L., Jr. Metagenomic Analysis of the Respiratory Microbiome of a Broiler Flock from Hatching to Processing. Microorganisms 2021, 9, 721. https://doi.org/10.3390/microorganisms9040721
Mulholland KA, Robinson MG, Keeler SJ, Johnson TJ, Weber BW, Keeler CL Jr. Metagenomic Analysis of the Respiratory Microbiome of a Broiler Flock from Hatching to Processing. Microorganisms. 2021; 9(4):721. https://doi.org/10.3390/microorganisms9040721
Chicago/Turabian StyleMulholland, Kelly A., Monique G. Robinson, Sharon J. Keeler, Timothy J. Johnson, Bonnie W. Weber, and Calvin L. Keeler, Jr. 2021. "Metagenomic Analysis of the Respiratory Microbiome of a Broiler Flock from Hatching to Processing" Microorganisms 9, no. 4: 721. https://doi.org/10.3390/microorganisms9040721