
Active Rumen Bacterial and Protozoal Communities Revealed by RNA-Based Amplicon Sequencing on Dairy Cows Fed Different Diets at Three Physiological Stages




Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal Management, Diet, and Rumen Fluid Sampling
2.3. RNA-Extraction and Synthesis of cDNA
2.4. Targeted Amplicon Sequencing of Bacteria and Protozoa
2.5. Bioinformatic Analysis
2.6. Statistical Analysis
3. Results
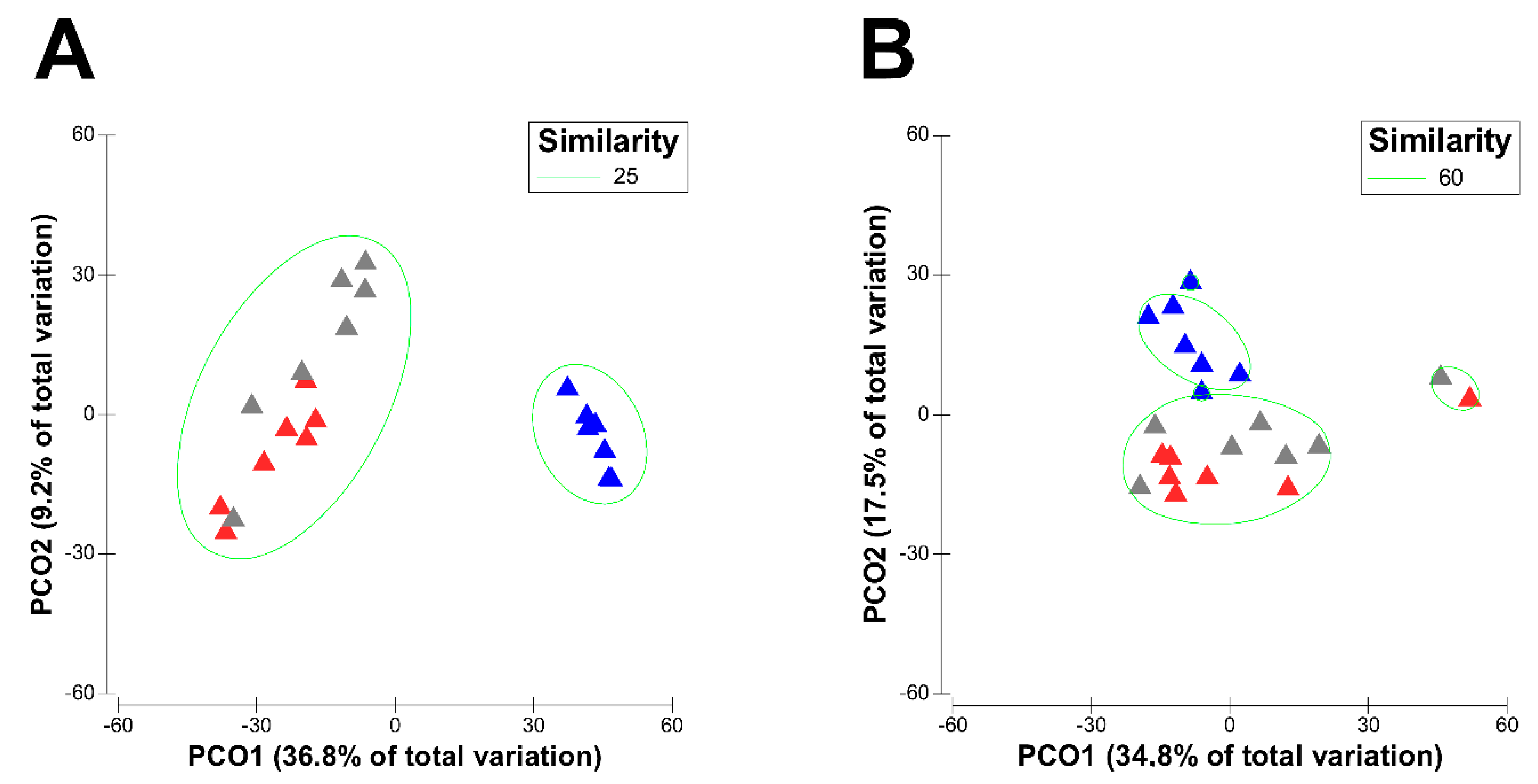
3.1. Effect of Physiological Stages of Dairy Cows on the Diversity of Their Active Rumen Microbiota
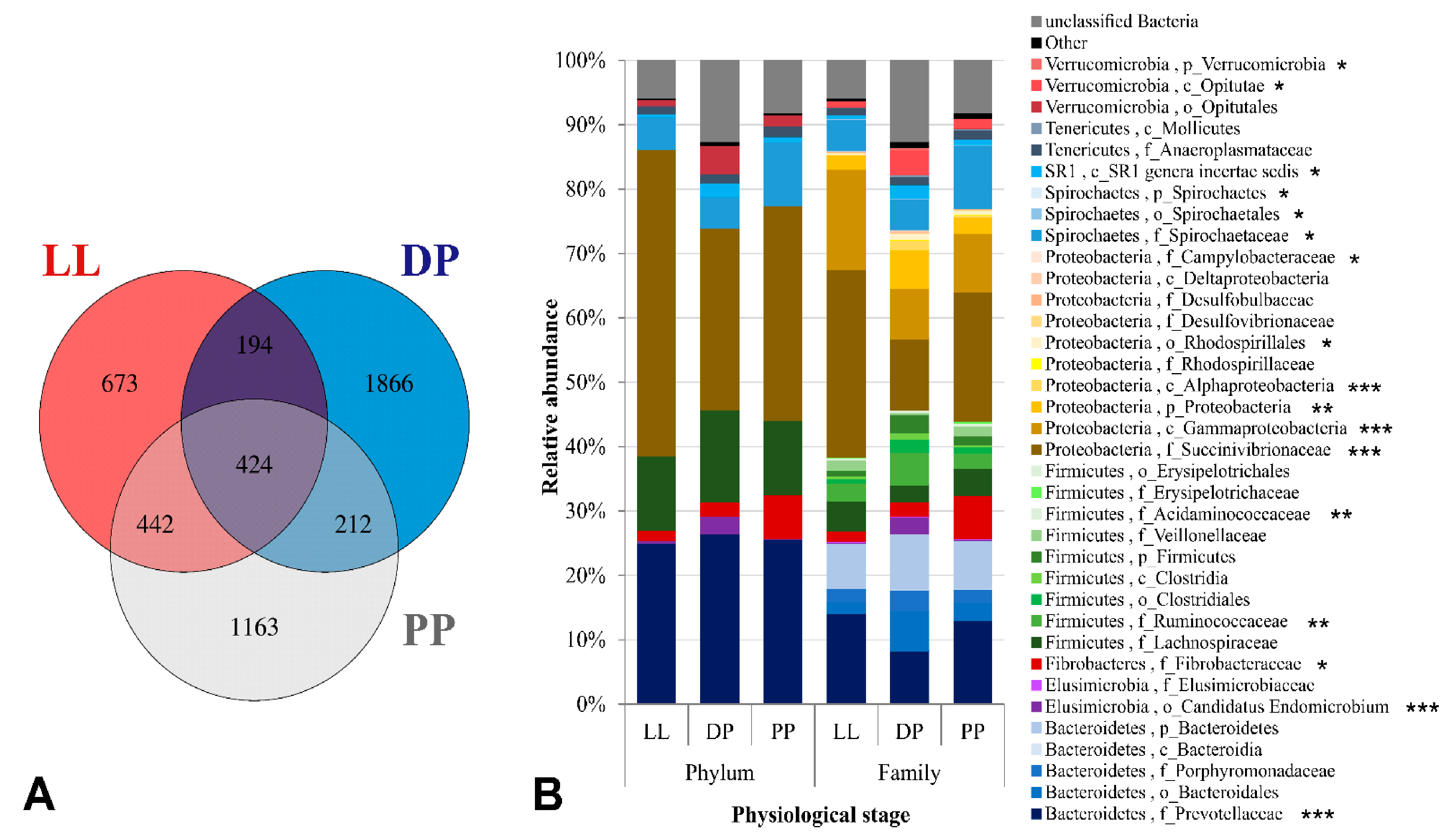
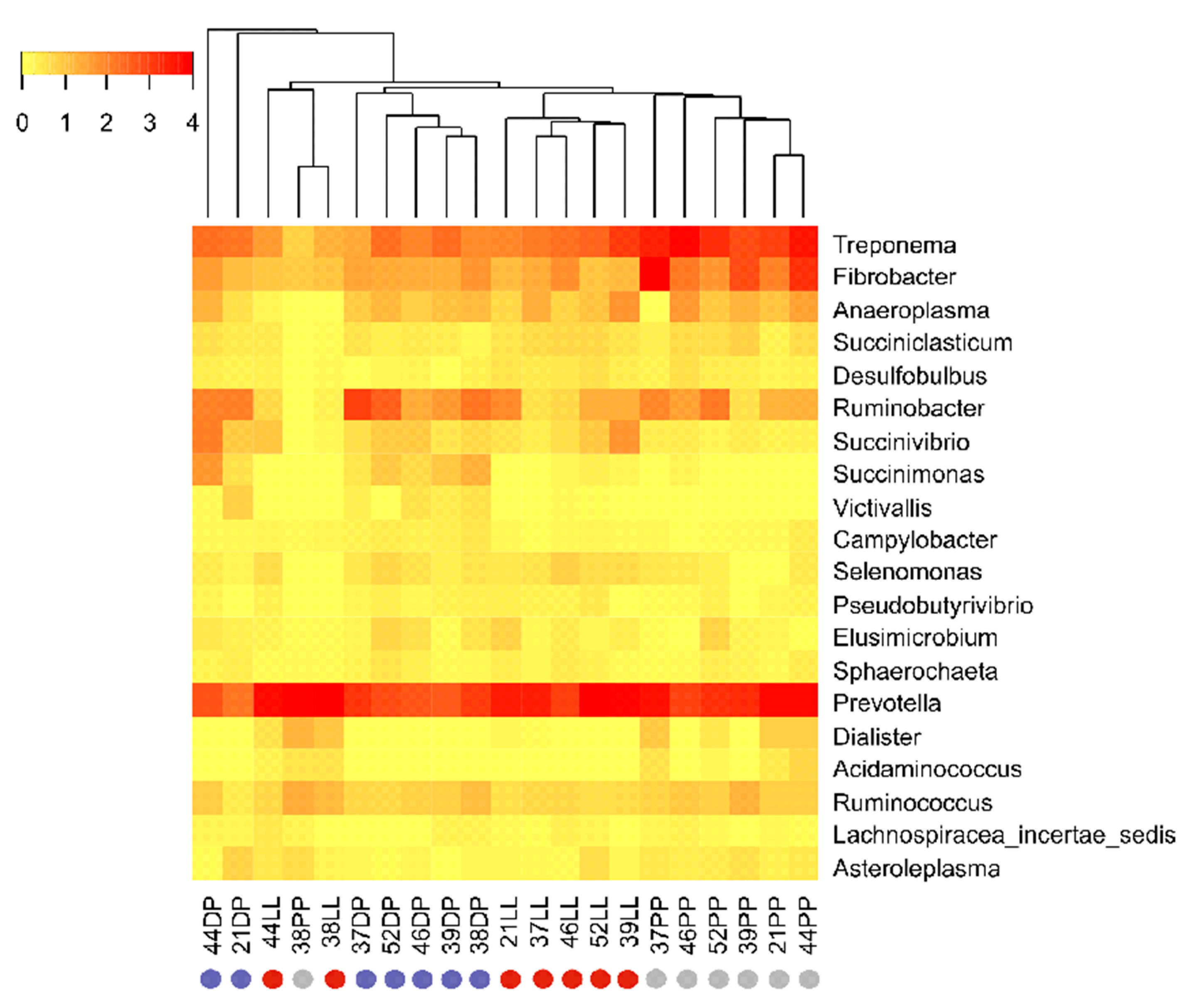
3.2. Physiological Stage-Dependent Modifications in the Active Rumen Bacterial Communities
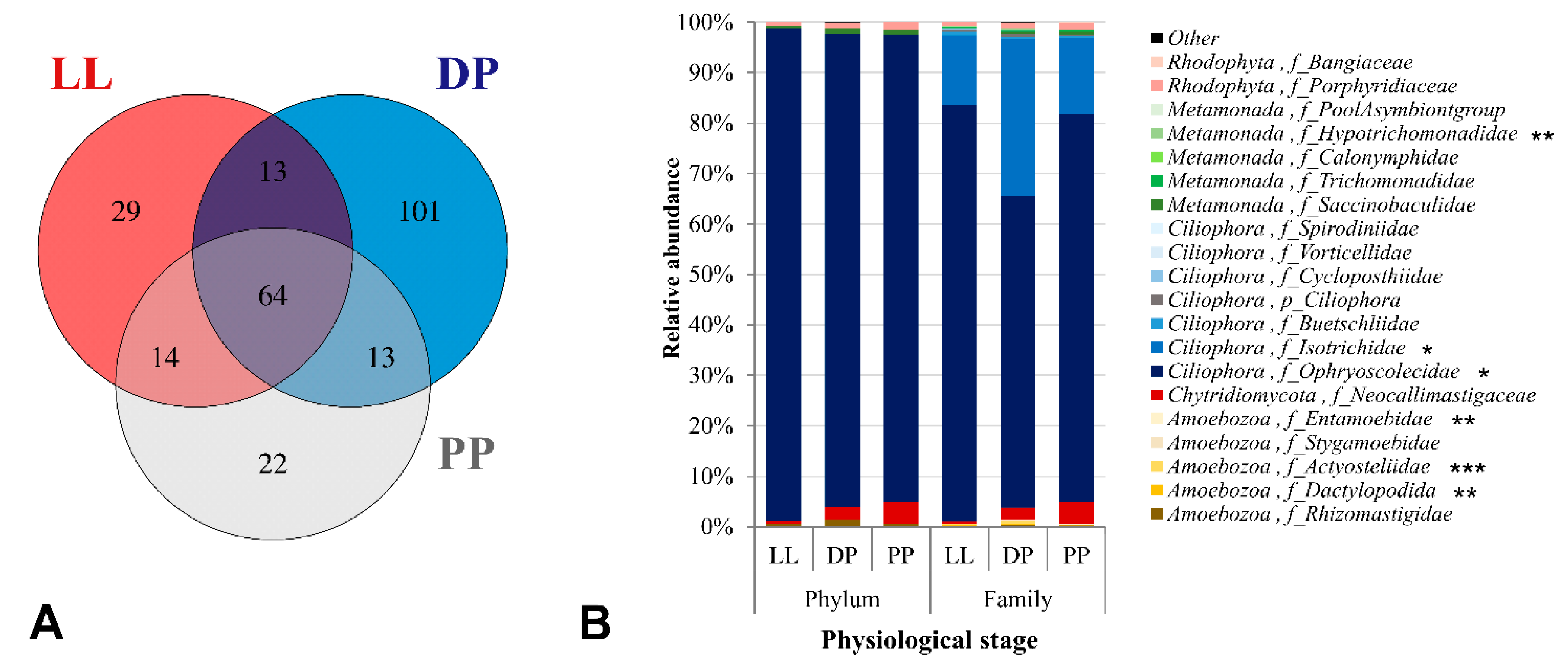
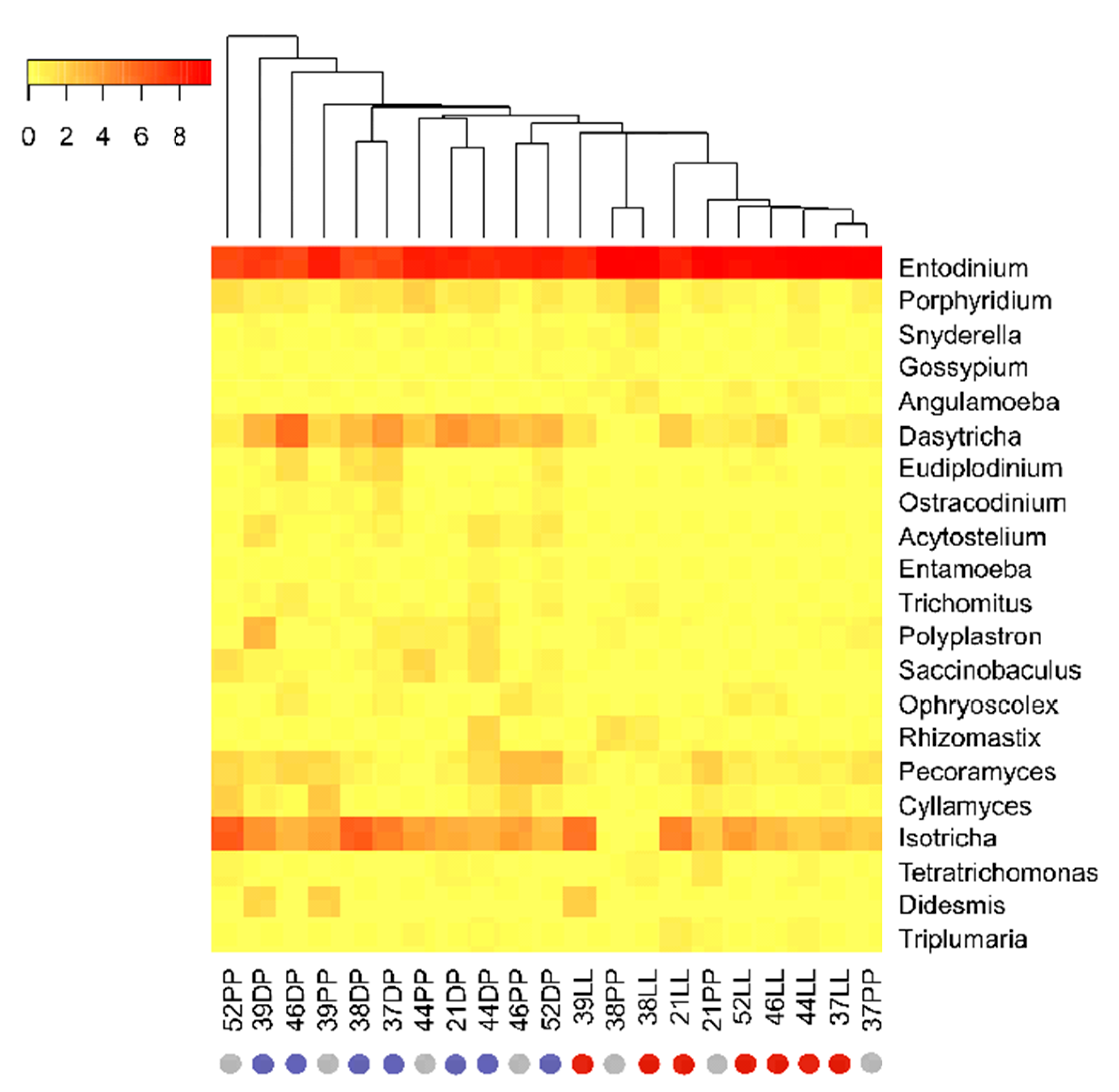
3.3. Physiological Stage-Dependent Modifications in the Active Rumen Eukaryotic Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, W.; Xi, D.; Mao, H.; Wanapat, M. The use of molecular techniques based on ribosomal RNA and DNA for rumen microbial ecosystem studies: A review. Mol. Biol. Rep. 2008, 35, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Shabat, S.K.B.; Sasson, G.; Doron-Faigenboim, A.; Durman, T.; Yaacoby, S.; Miller, M.E.B.; White, B.A.; Shterzer, N.; Mizrahi, I. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef] [PubMed]
- Petri, R.M.; Schwaiger, T.; Penner, G.B.; Beauchemin, K.A.; Forster, R.J.; McKinnon, J.J.; McAllister, T.A. Characterization of the core rumen microbiome in cattle during transition from forage to concentrate as well as during and after an acidotic challenge. PLoS ONE 2013, 8, e83424. [Google Scholar] [CrossRef]
- Zened, A.; Combes, S.; Cauquil, L.; Mariette, J.; Klopp, C.; Bouchez, O.; Troegeler-Meynadier, A.; Enjalbert, F. Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiol. Ecol. 2013, 83, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, M.H.; Tahmasbi, A.M.; Khorvash, M.; Naserian, A.A.; Ghaffari, A.H.; Valizadeh, H. Effects of pistachio by-products in replacement of alfalfa hay on populations of rumen bacteria involved in biohydrogenation and fermentative parameters in the rumen of sheep. J. Anim. Physiol. Anim. Nutr. 2014, 98, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Penner, G.B.; Hernandez-Sanabria, E.; Oba, M.; Guan, L.L. Effects of sampling location and time, and host animal on assessment of bacterial diversity and fermentation parameters in the bovine rumen. J. Appl. Microbiol. 2009, 107, 1924–1934. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Paz, H.A.; Anderson, C.L.; Muller, M.J.; Kononoff, P.J.; Fernando, S.C. Rumen bacterial community composition in holstein and jersey cows is different under same dietary condition and is not affected by sampling method. Front. Microbiol. 2016, 7, 1206. [Google Scholar] [CrossRef]
- Furman, O.; Shenhav, L.; Sasson, G.; Kokou, F.; Honig, H.; Jacoby, S.; Hertz, T.; Cordero, O.; Halperin, E.; Mizrahi, I. Stochasticity constrained by deterministic effects of diet and age drive rumen microbiome assembly dynamics. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Abbas, W.; Howard, J.T.; Paz, H.A.; Hales, K.E.; Wells, J.E.; Kuehn, L.A.; Erickson, G.E.; Spangler, M.L.; Fernando, S.C. Influence of host genetics in shaping the rumen bacterial community in beef cattle. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Gruninger, R.J.; Ribeiro, G.O.; Cameron, A.; McAllister, T.A. Invited review: Application of meta-omics to understand the dynamic nature of the rumen microbiome and how it responds to diet in ruminants. Animal 2019, 13, 1843–1854. [Google Scholar] [CrossRef]
- Auffret, M.D.; Dewhurst, R.J.; Duthie, C.A.; Rooke, J.A.; Wallace, R.J.; Freeman, T.C.; Stewart, R.; Watson, M.; Roehe, R. The rumen microbiome as a reservoir of antimicrobial resistance and pathogenicity genes is directly affected by diet in beef cattle. Microbiome 2017, 5, 159. [Google Scholar] [CrossRef]
- Greter, A.M.; DeVries, T.J.; Von Keyserlingk, M.A.G. Nutrient intake and feeding behavior of growing dairy heifers: Effects of dietary dilution. J. Dairy Sci. 2008, 91, 2786–2795. [Google Scholar] [CrossRef]
- Grum, D.E.; Drackley, J.K.; Hansen, L.R.; Cremin, J.D., Jr. Production, Digestion, and Hepatic Lipid Metabolism of Dairy Cows Fed Increased Energy from Fat or Concentrate. J. Dairy Sci. 1996, 79, 1836–1849. [Google Scholar] [CrossRef]
- Coblentz, W.K.; Esser, N.M.; Hoffman, P.C.; Akins, M.S. Growth performance and sorting characteristics of corn silage-alfalfa haylage diets with or without forage dilution offered to replacement Holstein dairy heifers. J. Dairy Sci. 2015, 98, 8018–8034. [Google Scholar] [CrossRef]
- Su, H.; Akins, M.S.; Esser, N.M.; Ogden, R.; Coblentz, W.K.; Kalscheur, K.F.; Hatfield, R. Effects of feeding alfalfa stemlage or wheat straw for dietary energy dilution on nutrient intake and digestibility, growth performance, and feeding behavior of Holstein dairy heifers. J. Dairy Sci. 2017, 100, 7106–7115. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, Y.; Xu, Q.; Zheng, N.; Zhao, S.; Huang, G.; Wang, J. Ruminal microbiota-host interaction and its effect on nutrient metabolism. Anim. Nutr. 2020, 7, 49–55. [Google Scholar] [CrossRef]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef]
- Cersosimo, L.M.; Bainbridge, M.L.; Wright, A.D.G.; Kraft, J. Breed and lactation stage alter the rumen protozoal fatty acid profiles and community structures in primiparous dairy cattle. J. Agric. Food Chem. 2016, 64, 2021–2029. [Google Scholar] [CrossRef]
- Wang, Y.; Nan, X.; Zhao, Y.; Wang, Y.; Jiang, L.; Xiong, B. Ruminal degradation of rumen-protected glucose influences ruminal microbiota and metabolites in early lactation dairy cows. Appl. Environ. Microbiol. 2020, 87. [Google Scholar] [CrossRef]
- Martinez-Fernandez, G.; Jiao, J.; Padmanabha, J.; Denman, S.E.; McSweeney, C.S. Seasonal and Nutrient Supplement Responses in Rumen Microbiota Structure and Metabolites of Tropical Rangeland Cattle. Microorganisms 2020, 8, 1550. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Neves, A.L.; Ghoshal, B. Symposium review: Mining metagenomic and metatranscriptomic data for clues about microbial metabolic functions in ruminants. J. Dairy Sci. 2018, 101, 5605–5618. [Google Scholar] [CrossRef] [PubMed]
- Pers-Kamczyc, E.; Zmora, P.; Cieslak, A.; Szumacher-Strabel, M. Development of nucleic acid based techniques and possibilities of their application to rumen microbial ecology research. J. Anim. Feed Sci. 2011, 20, 315–337. [Google Scholar] [CrossRef]
- Guzman, C.E.; Bereza-Malcolm, L.T.; De Groef, B.; Franks, A.E. Presence of selected methanogens, fibrolytic bacteria, and proteobacteria in the gastrointestinal tract of neonatal dairy calves from birth to 72 hours. PLoS ONE 2015, 10, e0133048. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Zhang, M.L.; Zhang, R.Y.; Zhu, W.Y.; Mao, S.Y. Comparative studies of the composition of bacterial microbiota associated with the ruminal content, ruminal epithelium and in the faeces of lactating dairy cows. Microb. Biotechnol. 2016, 9, 257–268. [Google Scholar] [CrossRef]
- Wang, L.; Xu, Q.; Kong, F.; Yang, Y.; Wu, D.; Mishra, S.; Li, Y. Exploring the goat rumen microbiome from seven days to two years. PLoS ONE 2016, 11, e0154354. [Google Scholar] [CrossRef]
- Gaidos, E.; Rusch, A.; Ilardo, M. Ribosomal tag pyrosequencing of DNA and RNA from benthic coral reef microbiota: Community spatial structure, rare members and nitrogen-cycling guilds. Environ. Microbiol. 2011, 13, 1138–1152. [Google Scholar] [CrossRef]
- Sessitsch, A.; Gyamfi, S.; Stralis-Pavese, N.; Weilharter, A.; Pfeifer, U. RNA isolation from soil for bacterial community and functional analysis: Evaluation of different extraction and soil conservation protocols. J. Microbiol. Methods 2002, 51, 171–179. [Google Scholar] [CrossRef]
- Keer, J.T.; Birch, L. Molecular methods for the assessment of bacterial viability. J. Microbiol. Methods 2003, 53, 175–183. [Google Scholar] [CrossRef]
- Kang, S.; Denman, S.E.; Morrison, M.; Yu, Z.; McSweeney, C.S. An efficient RNA extraction method for estimating gut microbial diversity by polymerase chain reaction. Curr. Microbiol. 2009, 58, 464–471. [Google Scholar] [CrossRef]
- Lee, S.; Kemp, P.F. Single-cell RNA content of natural marine planktonic bacteria measured by hybridization with multiple 16S rRNA-targeted fluorescent probes. Limnol. Oceanogr. 1994, 39, 869–879. [Google Scholar] [CrossRef]
- Kang, S.H.; Evans, P.; Morrison, M.; Mcsweeney, C. Identification of metabolically active proteobacterial and archaeal communities in the rumen by DNA- and RNA-derived 16S rRNA gene. J. Appl. Microbiol. 2013, 115, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Lettat, A.; Hassanat, F.; Benchaar, C. Corn silage in dairy cow diets to reduce ruminal methanogenesis: Effects on the rumen metabolically active microbial communities. J. Dairy Sci. 2013, 96, 5237–5248. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Henderson, G.; Sun, X.; Cox, F.; Janssen, P.H.; Guan, L.L. Taxonomic assessment of rumen microbiota using total rna and targeted amplicon sequencing approaches. Front. Microbiol. 2016, 7, 987. [Google Scholar] [CrossRef]
- Tagliapietra, F.; Cattani, M.; Hindrichsen, I.K.; Hansen, H.H.; Colombini, S.; Bailoni, L.; Schiavon, S. True dry matter digestibility of feeds evaluated in situ with different bags and in vitro using rumen fluid collected from intact donor cows. Anim. Prod. Sci. 2012, 52, 338–346. [Google Scholar] [CrossRef]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef]
- Amaral-Zettler, L.A.; McCliment, E.A.; Ducklow, H.W.; Huse, S.M. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA Genes. PLoS ONE 2009, 4, e6372. [Google Scholar] [CrossRef]
- Stoeck, T.; Bass, D.; Nebel, M.; Christen, R.; Jones, M.D.M.; Breiner, H.W.; Richards, T.A. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 2010, 19, 21–31. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Cole, J.R.; Chai, B.; Farris, R.J.; Wang, Q.; Kulam, S.A.; McGarrell, D.M.; Garrity, G.M.; Tiedje, J.M. The Ribosomal Database Project (RDP-II): Sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 2005, 33, D294–D296. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef]
- Anderson, M.J.; Gorley, R.N.; Clarke, K.R. PERMANOVA+ for PRIMER. Guide to Software and Statistical Methods; PRIMER-E Ltd.: Plymouth, UK, 2008. [Google Scholar]
- Lima, F.S.; Oikonomou, G.; Lima, S.F.; Bicalho, M.L.S.; Ganda, E.K.; de Oliveira Filho, J.C.; Lorenzo, G.; Trojacanec, P.; Bicalho, R.C. Prepartum and postpartum rumen fluid microbiomes: Characterization and correlation with production traits in dairy cows. Appl. Environ. Microbiol. 2015, 81, 1327–1337. [Google Scholar] [CrossRef]
- Zhu, Z.; Noel, S.J.; Difford, G.F.; Al-Soud, W.A.; Brejnrod, A.; Sørensen, S.J.; Lassen, J.; Løvendahl, P.; Højberg, O. Community structure of the metabolically active rumen bacterial and archaeal communities of dairy cows over the transition period. PLoS ONE 2017, 12, e0187858. [Google Scholar] [CrossRef]
- Liu, J.; Tian, K.; Sun, Y.; Wu, Y.; Chen, J.; Zhang, R.; Dong, G. Effects of the acid–base treatment of corn on rumen fermentation and microbiota, inflammatory response and growth performance in beef cattle fed high-concentrate diet. Animal 2020, 14, 1876–1884. [Google Scholar] [CrossRef]
- Belanche, A.; De la Fuente, G.D.; Moorby, J.M.; Newbold, C.J. Bacterial protein degradation by different rumen protozoal groups. J. Anim. Sci. 2012, 90, 4495–4504. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, H.; Wang, Y.; Li, S.; Cao, Z.; Ji, S.; He, Y.; Zhang, H. Effect of dietary forage to concentrate ratios on dynamic profile changes and interactions of ruminal microbiota and metabolites in Holstein heifers. Front. Microbiol. 2017, 8, 2206. [Google Scholar] [CrossRef]
- Pinto, A.C.; Bertoldi, G.P.; Felizari, L.D.; Dias, E.F.; Demartini, B.L.; Nunes, A.B.; Squizatti, M.; Silvestre, A.; Oliveira, L.; Skarlupka, J.; et al. Ruminal fermentation pattern, bacterial community composition, and nutrient digestibility of Nellore cattle submitted to either nutritional restriction or intake of concentrate feedstuffs prior to adaptation period. Front. Microbiol. 2020, 11, 1865. [Google Scholar] [CrossRef]
- Dehority, B.A.; Odenyo, A.A. Influence of diet on the rumen protozoal fauna of indigenous African wild ruminants. J. Eukaryot. Microbiol. 2003, 50, 220–223. [Google Scholar] [CrossRef]
- Tymensen, L.; Barkley, C.; McAllister, T.A. Relative diversity and community structure analysis of rumen protozoa according to T-RFLP and microscopic methods. J. Microbiol. Methods 2012, 88, 1–6. [Google Scholar] [CrossRef]
- Li, F.; Guan, L. Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Yu, Z. Variations in 16S rRNA-based microbiome profiling between pyrosequencing runs and between pyrosequencing facilities. J. Microbiol. 2014, 52, 355–365. [Google Scholar] [CrossRef]
- Auffret, M.D.; Stewart, R.; Dewhurst, R.J.; Duthie, C.A.; Rooke, J.A.; Wallace, R.J.; Freeman, T.C.; Snelling, T.J.; Watson, M.; Roehe, R. Identification, comparison, and validation of robust rumen microbial biomarkers for methane emissions using diverse Bos Taurus breeds and basal diets. Front. Microbiol. 2018, 8, 2642. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Kittelmann, S.; Miri, V.H.; Zethof, M.; Noel, S.J.; Waghorn, G.C.; Janssen, P.H. Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities. PLoS ONE 2013, 8, e74787. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 1–15. [Google Scholar] [CrossRef]
- Russell, J.B.; Rychlik, J.L. Factors that alter rumen microbial ecology. Science 2001, 292, 1119–1122. [Google Scholar] [CrossRef]
- Liu, Y.; Whitman, W.B. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N. Y. Acad. Sci. 2008, 1125, 171–189. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.S.; Cormican, P.; Keogh, K.; O’Connor, A.; O’Hara, E.; Palladino, R.A.; Kenny, D.A.; Waters, S.M. Illumina MiSeq phylogenetic amplicon sequencing shows a large reduction of an uncharacterised succinivibrionaceae and an increase of the Methanobrevibacter gottschalkii clade in feed restricted cattle. PLoS ONE 2015, 10, e0133234. [Google Scholar] [CrossRef] [PubMed]
- Pope, P.B.; Smith, W.; Denman, S.E.; Tringe, S.G.; Barry, K.; Hugenholtz, P.; McSweeney, C.S.; McHardy, A.C.; Morrison, M. Isolation of Succinivibrionaceae implicated in low methane emissions from Tammar wallabies. Science 2011, 333, 646–648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Liu, J.; Jiang, L.; Mao, S. Effect of high-concentrate diets on microbial composition, function, and the VFAs formation process in the rumen of dairy cows. Anim. Feed Sci. Technol. 2020, 269, 114619. [Google Scholar] [CrossRef]
- Van Gylswyk, N.O. Enumeration and presumptive identification of some functional groups of bacteria in the rumen of dairy cows fed grass silage-based diets. FEMS Microbiol. Ecol. 1990, 6, 243–261. [Google Scholar] [CrossRef]
- Purushe, J.; Fouts, D.E.; Morrison, M.; White, B.A.; Mackie, R.I.; Coutinho, P.M.; Henrissat, B.; Nelson, K.; North American Consortium for Rumen Bacteria. Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: Insights into their environmental niche. Microb. Ecol. 2010, 60, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Ushida, K.; Miyazaki, K.; Kojima, Y. Use of ratio of digested xylan to digested cellulose (X/C) as an index of fiber digestion in plant cell-wall material by ruminal microorganisms. Anim. Feed Sci. Technol. 1998, 71, 207–215. [Google Scholar] [CrossRef]
- Attwood, G.T.; Reilly, K. Identification of proteolytic rumen bacteria isolated from New Zealand cattle. J. Appl. Bacteriol. 1995, 79, 22–29. [Google Scholar] [CrossRef]
- Kljak, K.; Pino, F.; Harvatine, K.J.; Heinrichs, A.J. Analysis of selected rumen microbial populations in dairy heifers limit fed diets varying in trace mineral form and starch content. Livest. Sci. 2017, 198, 93–96. [Google Scholar] [CrossRef]
- Suen, G.; Stevenson, D.M.; Bruce, D.C.; Chertkov, O.; Copeland, A.; Cheng, J.F.; Detter, C.; Detter, J.C.; Goodwin, L.A.; Han, C.S.; et al. Complete genome of the cellulolytic ruminal bacterium Ruminococcus albus 7. J. Bacteriol. 2011, 193, 5574–5575. [Google Scholar] [CrossRef]
- Wang, L.; Hatem, A.; Catalyurek, U.V.; Morrison, M.; Yu, Z. Metagenomic Insights into the Carbohydrate-Active Enzymes Carried by the Microorganisms Adhering to Solid Digesta in the Rumen of Cows. PLoS ONE 2013, 8, e78507. [Google Scholar] [CrossRef]
- Huws, S.A.; Kim, E.J.; Lee, M.R.F.; Scott, M.B.; Tweed, J.K.S.; Pinloche, E.; Wallace, R.J.; Scollan, N.D. As yet uncultured bacteria phylogenetically classified as Prevotella, Lachnospiraceae incertae sedis and unclassified Bacteroidales, Clostridiales and Ruminococcaceae may play a predominant role in ruminal biohydrogenation. Environ. Microbiol. 2011, 13, 1500–1512. [Google Scholar] [CrossRef]
- Zhao, L.; Meng, Q.; Ren, L.; Liu, W.; Zhang, X.; Huo, Y.; Zhou, Z. Effects of nitrate addition on rumen fermentation, bacterial biodiversity and abundance. Asian-Australas. J. Anim. Sci. 2015, 28, 1433. [Google Scholar] [CrossRef]
- Lourenco, J.M.; Kieran, T.J.; Seidel, D.S.; Glenn, T.C.; Silveira, M.F.D.; Callaway, T.R.; Stewart, R.L., Jr. Comparison of the ruminal and fecal microbiotas in beef calves supplemented or not with concentrate. PLoS ONE 2020, 15, e0231533. [Google Scholar] [CrossRef]
- Shin, E.C.; Choi, B.R.; Lim, W.J.; Hong, S.Y.; An, C.L.; Cho, K.M.; Kim, Y.K.; An, J.M.; Kang, J.M.; Lee, S.S.; et al. Phylogenetic analysis of archaea in three fractions of cow rumen based on the 16S rDNA sequence. Anaerobe 2004, 10, 313–319. [Google Scholar] [CrossRef]
- Leng, J.; Zhong, X.; Zhu, R.J.; Yang, S.L.; Gou, X.; Mao, H.M. Assessment of protozoa in Yunnan Yellow Cattle rumen based on the 18S rRNA sequences. Mol. Biol. Rep. 2011, 38, 577–585. [Google Scholar] [CrossRef]
- Benchaar, C.; Hassanat, F.; Gervais, R.; Chouinard, P.Y.; Petit, H.V.; Massé, D. Methane production, digestion, ruminal fermentation, nitrogen balance, and milk production of cows fed corn silage- or barley silage-based diets. J. Dairy Sci. 2014, 97, 961–974. [Google Scholar] [CrossRef]
- Iqbal, M.W.; Zhang, Q.; Yang, Y.; Li, L.; Zou, C.; Huang, C.; Lin, B. Comparative study of rumen fermentation and microbial community differences between water buffalo and Jersey cows under similar feeding conditions. J. Appl. Anim. Res. 2018, 46, 740–748. [Google Scholar] [CrossRef]
- Matthews, C.; Crispie, F.; Lewis, E.; Reid, M.; O’Toole, P.W.; Cotter, P.D. The rumen microbiome: A crucial consideration when optimising milk and meat production and nitrogen utilisation efficiency. Gut Microbes 2019, 10, 115–132. [Google Scholar] [CrossRef]
- Williams, A.G.; Coleman, G.S. The Rumen Protozoa, 1st ed.; Springer: New York, NY, USA, 1992; ISBN 978-1-4612-7664-7. [Google Scholar]
- Karnati, S.K.R.; Yu, Z.; Sylvester, J.T.; Dehority, B.A.; Morrison, M.; Firkins, J.L. Technical note: Specific PCR amplification of protozoal 18S rDNA sequences from DNA extracted from ruminal samples of cows. J. Anim. Sci. 2003, 81, 812–815. [Google Scholar] [CrossRef]
- Ueda, K.; Ferlay, A.; Chabrot, J.; Loor, J.J.; Chilliard, Y.; Doreau, M. Effect of linseed oil supplementation on ruminal digestion in dairy cows fed diets with different forage:concentrate ratios. J. Dairy Sci. 2003, 86, 3999–4007. [Google Scholar] [CrossRef]
- Messana, J.D.; Carvalho, A.L.E.G.F.; Ribeiro, A.F.; Fiorentini, G.; Castagnino, P.S.; Granja-Salcedo, Y.T.; Pires, A.V.; Berchielli, T.T. Effects of different sources of forage in high-concentrate diets on fermentation parameters, ruminal biohydrogenation and microbiota in Nellore feedlot steers. J. Agric. Sci. 2016, 154, 928–941. [Google Scholar] [CrossRef]
- Orpin, C.G.; Letcher, A.J. Effect of absence of ciliate protozoa on rumen fluid volume, flow rate and bacterial populations in sheep. Anim. Feed Sci. Technol. 1984, 10, 145–153. [Google Scholar] [CrossRef]
- Newbold, C.J.; De La Fuente, G.; Belanche, A.; Ramos-Morales, E.; McEwan, N.R. The role of ciliate protozoa in the rumen. Front. Microbiol. 2015, 6, 1313. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.L.; Thomas, B.J.; McEwan, N.R.; Rees Stevens, P.; Creevey, C.J.; Huws, S.A. Rumen protozoa play a significant role in fungal predation and plant carbohydrate breakdown. Front. Microbiol. 2020, 11, 720. [Google Scholar] [CrossRef] [PubMed]
- Coleman, G.S. The rate of uptake and metabolism of starch grains and cellulose particles by Entodinium species, Eudiplodinium maggii, some other entodiniomorphid protozoa and natural protozoal populations taken from the ovine rumen. J. Appl. Bacteriol. 1992, 73, 507–513. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Physiological Stage 1 | ||
|---|---|---|---|
| LL | DP | PP | |
| Ingredients | |||
| Meadow hay | 13.1 | 24.6 | 2.5 |
| Alfalfa hay | 17.4 | - | 20.6 |
| Maize silage | 47.4 | 20.3 | 32.0 |
| Wheat straw | - | 37.8 | 1.7 |
| Dry sugar beet pulp | - | - | 2.4 |
| Protein mix 2 | 10.6 | 16.1 | 17.7 |
| Energy mix 3 | 8.9 | - | 19.3 |
| Extruded linseed | 1.1 | - | 0.4 |
| Fat supplement 4 | - | - | 1.2 |
| Vit–min mix 5,6 | 1.4 5 | 1.2 6 | 2.2 5 |
| Chemical composition | |||
| Dry matter | 50.2 | 63.8 | 55.1 |
| Crude protein | 13.1 | 13.0 | 15.7 |
| Lipids | 3.4 | 3.2 | 4.2 |
| Starch | 21.1 | 7.1 | 24.0 |
| NDF | 39.4 | 57.0 | 33.6 |
| ADF | 23.4 | 36.0 | 21.1 |
| MFU no./kg DM 7 | |||
| Index 1 | Bacteria | Eukaryotes | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LL | DP | PP | SEM 2 | p-Value 3 | LL | DP | PP | SEM 2 | p-Value 3 | |
| Richness | ||||||||||
| Chao 1 | 464 b | 692 a | 543 a,b | 36.50 | 0.026 | 47.0 b | 68.0 a | 40.0 b | 3.47 | <0.001 |
| Evenness | ||||||||||
| Shannon | 0.71 b | 0.84 a | 0.74 b | 0.02 | <0.001 | 0.39 | 0.48 | 0.39 | 0.03 | 0.3000 |
| Simpson | 0.93 b | 0.98 a | 0.96 a,b | 0.01 | 0.002 | 0.62 | 0.77 | 0.58 | 0.04 | 0.110 |
| Diversity | ||||||||||
| Shannon | 4.35 b | 5.50 a | 4.66 b | 0.14 | <0.001 | 1.45 a,b | 2.01 a | 1.43 b | 0.11 | 0.031 |
| Inverse Simpson | 16.4 b | 86.6 a | 32.4 a | 8.56 | <0.001 | 3.18 | 4.66 | 3.02 | 0.33 | 0.081 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bailoni, L.; Carraro, L.; Cardin, M.; Cardazzo, B. Active Rumen Bacterial and Protozoal Communities Revealed by RNA-Based Amplicon Sequencing on Dairy Cows Fed Different Diets at Three Physiological Stages. Microorganisms 2021, 9, 754. https://doi.org/10.3390/microorganisms9040754
Bailoni L, Carraro L, Cardin M, Cardazzo B. Active Rumen Bacterial and Protozoal Communities Revealed by RNA-Based Amplicon Sequencing on Dairy Cows Fed Different Diets at Three Physiological Stages. Microorganisms. 2021; 9(4):754. https://doi.org/10.3390/microorganisms9040754
Chicago/Turabian StyleBailoni, Lucia, Lisa Carraro, Marco Cardin, and Barbara Cardazzo. 2021. "Active Rumen Bacterial and Protozoal Communities Revealed by RNA-Based Amplicon Sequencing on Dairy Cows Fed Different Diets at Three Physiological Stages" Microorganisms 9, no. 4: 754. https://doi.org/10.3390/microorganisms9040754
APA StyleBailoni, L., Carraro, L., Cardin, M., & Cardazzo, B. (2021). Active Rumen Bacterial and Protozoal Communities Revealed by RNA-Based Amplicon Sequencing on Dairy Cows Fed Different Diets at Three Physiological Stages. Microorganisms, 9(4), 754. https://doi.org/10.3390/microorganisms9040754