
Differences in Gut Microbiome Composition and Antibiotic Resistance Gene Distribution between Chinese and Pakistani University Students from a Common Peer Group

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, PCR Amplification and MiSeq Sequencing
2.3. Bioinformatics Analysis
2.4. Quantification of ARGs
2.5. Statistical Analysis
3. Results
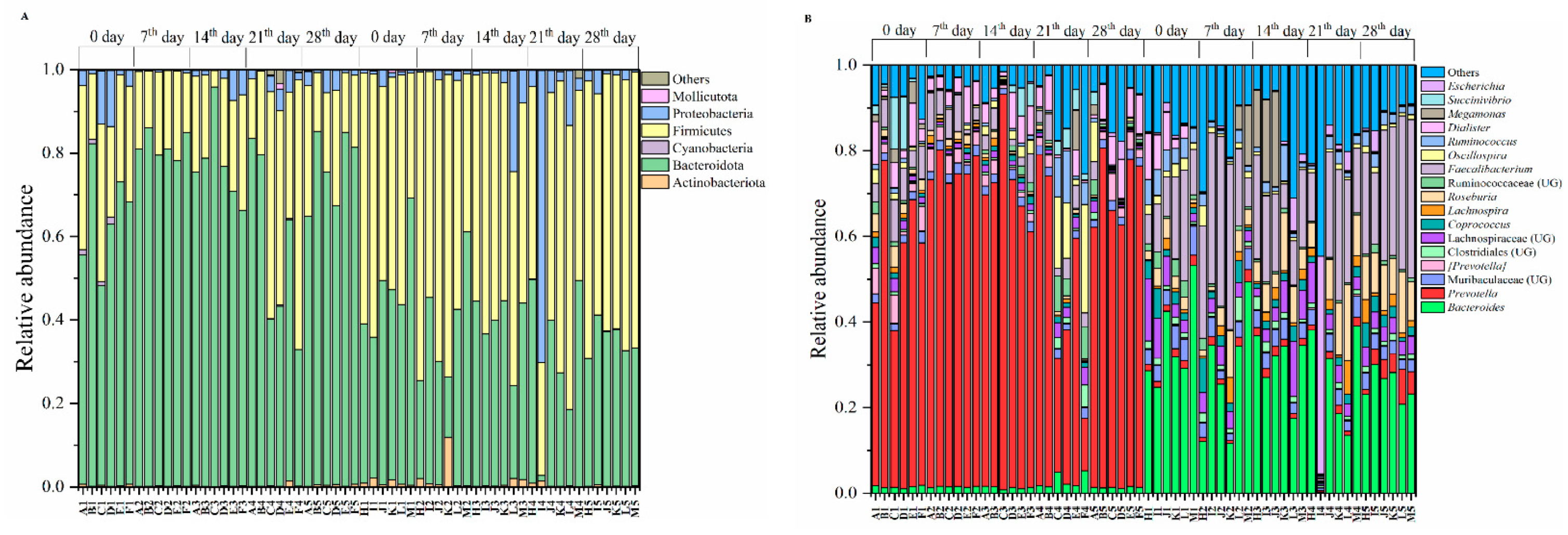
3.1. Exercise Is Not Influencing the Gut Microbiomes
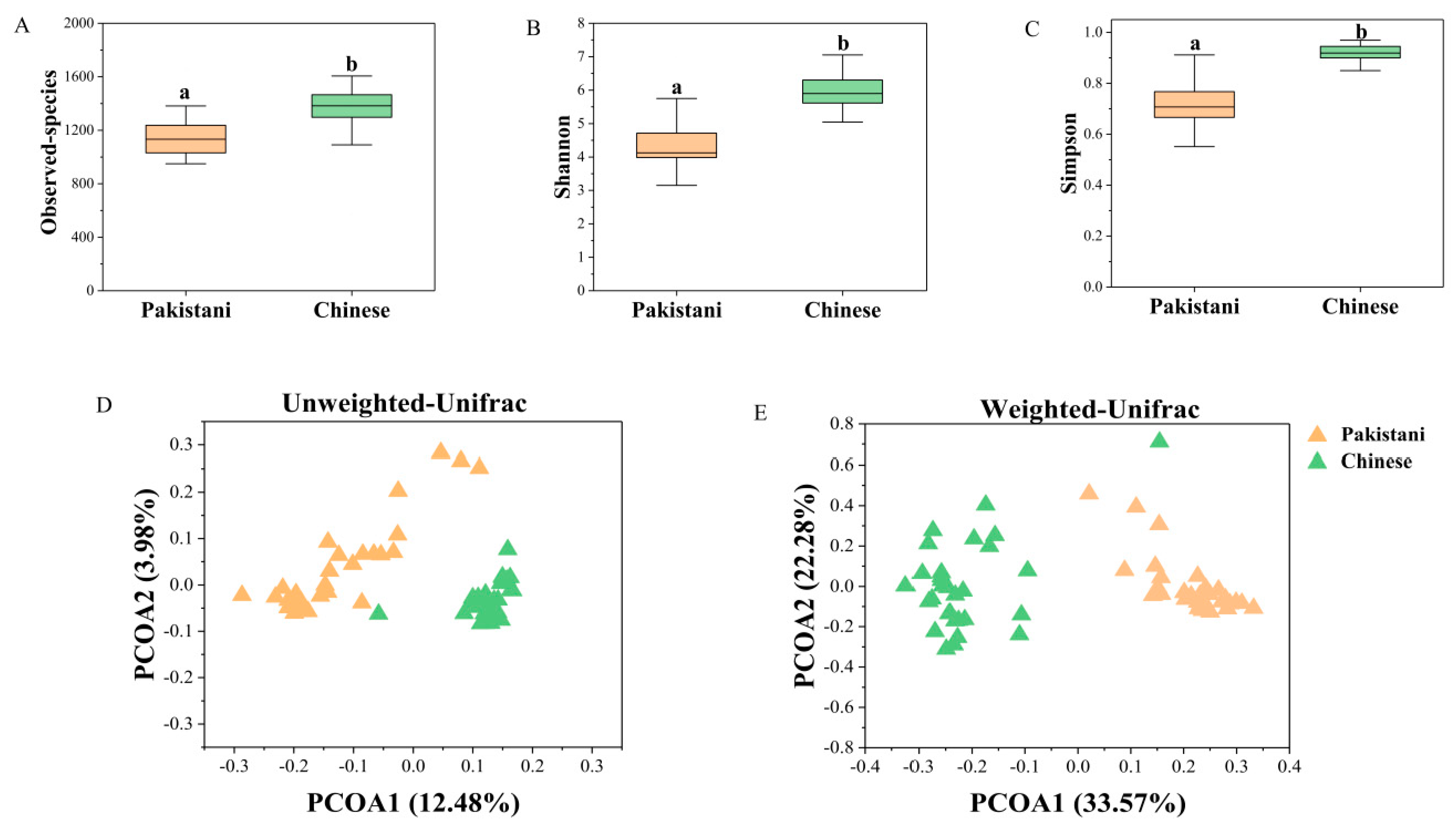
3.2. Gut Microbiomes Were Distributed Differently in Chinese and Pakistani Participants
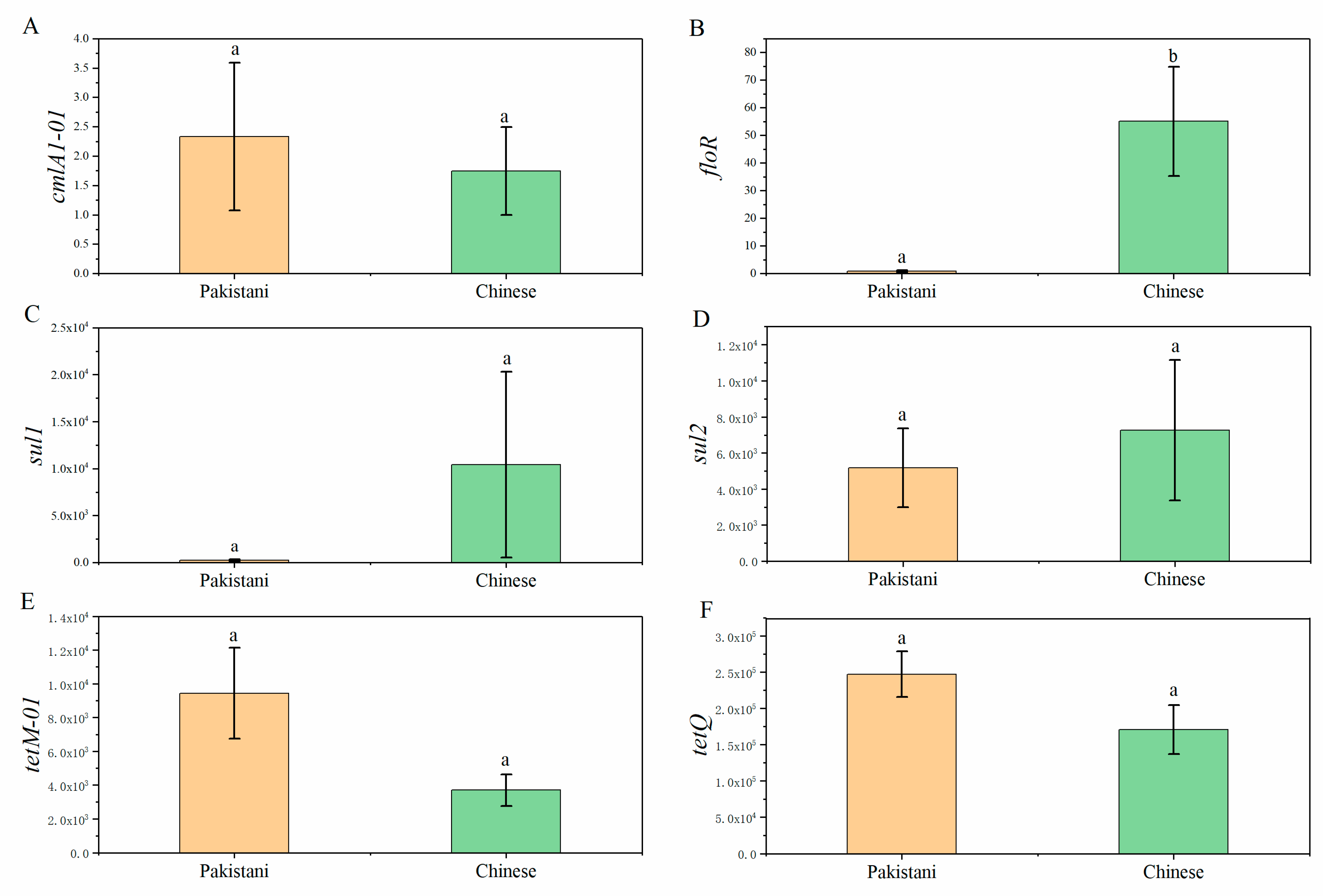
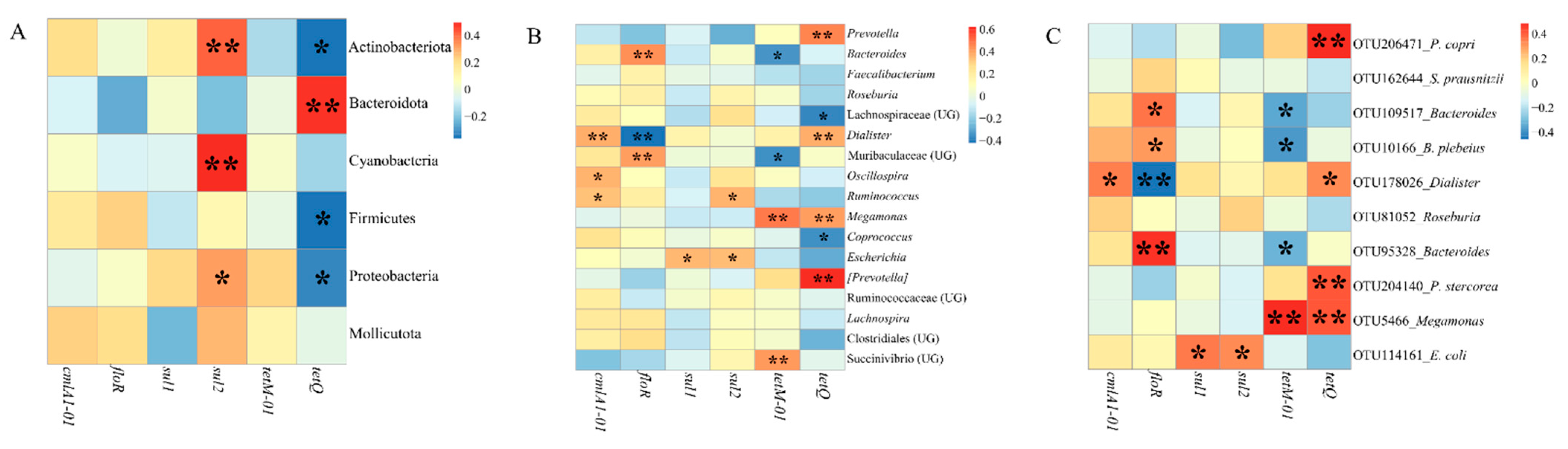
3.3. Distributions of Antibiotic Resistance Genes (ARGs) in Participants and Relationships between ARGs and Gut Microbiota
4. Discussion
4.1. Gut Microbiomes Remain Stable under the Intervention of Exercises during the Investigation Period
4.2. Different Nationalities Cause Variations of Gut Microbiomes
4.3. Some Dominant Gut Microbiomes Are Shared in People from Different Counties
4.4. Some Dominant Gut Microbiomes Have the Potential as Reservoirs of Antibiotic Resistance Genes (ARGs)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and Evolutionary Forces Shaping Microbial Diversity in the Human Intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Wang, X.; Li, L. Human gut microbiome: The second genome of human body. Protein Cell 2010, 1, 718–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmora, N.; Zeevi, D.; Korem, T.; Segal, E.; Elinav, E. Taking it Personally: Personalized Utilization of the Human Microbiome in Health and Disease. Cell Host Microbe 2016, 19, 12–20. [Google Scholar] [CrossRef]
- Butel, M.-J.; Waligora-Dupriet, A.-J.; Wydau-Dematteis, S. The developing gut microbiota and its consequences for health. J. Dev. Orig. Heal. Dis. 2018, 9, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.R.; Sanders, J.G.; Song, S.J.; Amato, K.R.; Clark, A.G.; Knight, R. The human microbiome in evolution. BMC Biol. 2017, 15, 1–12. [Google Scholar] [CrossRef]
- Hills, J.R.D.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut Microbiome: Profound Implications for Diet and Disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, C.A.T.N.Z.M.L.E. The microbiome and innate immunity. Nat. Cell Biol. 2016, 535, 65–74. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med Res. 2017, 4, 1–7. [Google Scholar] [CrossRef]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.J.; Sears, C.L.; Maruthur, N. Gut microbiome and its role in obesity and insulin resistance. Ann. New York Acad. Sci. 2019, 1461, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterol. 2020, 158, 322–340. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, W.; GEM Project Research Consortium; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Kevans, D.; I Smith, M.; Guttman, D.S.; Griffiths, A.; Panaccione, R.; et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 2016, 48, 1413–1417. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [Green Version]
- Dong, T.S.; Gupta, A. Influence of Early Life, Diet, and the Environment on the Microbiome. Clin. Gastroenterol. Hepatol. 2019, 17, 231–242. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nat. Cell Biol. 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Wilson, A.S.; Koller, K.R.; Ramaboli, M.C.; Nesengani, L.T.; Ocvirk, S.; Chen, C.; Flanagan, C.A.; Sapp, F.R.; Merritt, Z.T.; Bhatti, F.; et al. Diet and the Human Gut Microbiome: An International Review. Dig. Dis. Sci. 2020, 65, 723–740. [Google Scholar] [CrossRef] [Green Version]
- Bibbò, S.; Ianiro, G.; Giorgio, V.; Scaldaferri, F.; Masucci, L.; Gasbarrini, A.; Cammarota, G. The role of diet on gut microbiota composition. Eur. Rev. Med Pharmacol. Sci. 2016, 20, 4742–4749. [Google Scholar]
- Bermon, S.; Petriz, B.; Kajeniene, A.; Prestes, J.; Castell, L.; Franco, O.L. The microbiota: An exercise immunology perspective. Exerc. Immunol Rev. 2015, 21, 70–79. [Google Scholar]
- Ecerdá, B.; Epérez, M.; Pérez-Santiago, J.D.; Tornero-Aguilera, J.F.; Egonzález-Soltero, R.; Elarrosa, M. Gut Microbiota Modification: Another Piece in the Puzzle of the Benefits of Physical Exercise in Health? Front. Physiol. 2016, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, W.; Penney, N.C.; Cronin, O.; Garcia-Perez, I.; Molloy, M.G.; Holmes, E.; Shanahan, F.; Cotter, P.D.; O’Sullivan, O. The microbiome of professional athletes differs from that of more sedentary subjects in composition and particularly at the functional metabolic level. Gut Microbiota 2018, 67, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Grosicki, G.J.; Durk, R.P.; Bagley, J.R. Rapid gut microbiome changes in a world-class ultramarathon runner. Physiol. Rep. 2019, 7, e14313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aminov, R.I. The role of antibiotics and antibiotic resistance in nature. Environ. Microbiol. 2009, 11, 2970–2988. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance—the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef] [Green Version]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.; Schwarz, C.; Froese, D.G.; Zazula, G.D.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nat. Cell Biol. 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Yu, K.; Yang, R.; Zhang, Q.; Yang, Y.; Chen, E.; Lin, L.; Luan, T.; Chen, W.; Chen, B. Metagenomic characterization of antibiotic resistance genes in Antarctic soils. Ecotoxicol. Environ. Saf. 2019, 176, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Bartoloni, A.; Pallecchi, L.; Rodríguez, H.; Fernandez, C.; Mantella, A.; Bartalesi, F.; Strohmeyer, M.; Kristiansson, C.; Gotuzzo, E.; Paradisi, F.; et al. Antibiotic resistance in a very remote Amazonas community. Int. J. Antimicrob. Agents 2009, 33, 125–129. [Google Scholar] [CrossRef]
- Laxminarayan, R. Antibiotic effectiveness: Balancing conservation against innovation. Science 2014, 345, 1299–1301. [Google Scholar] [CrossRef] [Green Version]
- McInnes, R.S.; McCallum, G.E.; Lamberte, L.E.; van Schaik, W. Horizontal transfer of antibiotic resistance genes in the human gut microbiome. Curr. Opin. Microbiol. 2020, 53, 35–43. [Google Scholar] [CrossRef]
- Hu, X.; Zhou, Q.; Luo, Y. Occurrence and source analysis of typical veterinary antibiotics in manure, soil, vegetables and groundwater from organic vegetable bases, northern China. Environ. Pollut. 2010, 158, 2992–2998. [Google Scholar] [CrossRef]
- Heuer, H.; Schmitt, H.; Smalla, K. Antibiotic resistance gene spread due to manure application on agricultural fields. Curr. Opin. Microbiol. 2011, 14, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.L. Antibiotics and Antibiotic Resistance Genes in Natural Environments. Science 2008, 321, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Mah, M.W.; A Memish, Z. Antibiotic resistance. An impending crisis. Saudi Med J. 2000, 21, 1125–1129. [Google Scholar] [PubMed]
- Henriksson, P.J.G.; Rico, A.; Troell, M.; Klinger, D.H.; Buschmann, A.H.; Saksida, S.; Chadag, M.V.; Zhang, W. Unpacking factors influencing antimicrobial use in global aquaculture and their implication for management: A review from a systems perspective. Sustain. Sci. 2018, 13, 1105–1120. [Google Scholar] [CrossRef] [Green Version]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Pruden, A.; Pei, R.; Storteboom, H.; Carlson, K.H. Antibiotic Resistance Genes as Emerging Contaminants: Studies in Northern Colorado. Environ. Sci. Technol. 2006, 40, 7445–7450. [Google Scholar] [CrossRef]
- Yang, F.-X.; Mao, D.-Q.; Luo, Y.; Wang, Q.; Mu, Q.-H. Horizontal transfer of antibiotic resistance genes in the environment. Ying yong sheng tai xue bao J. Appl. Ecol. 2013, 24, 2993–3002. [Google Scholar]
- Saleem, A.F.; Ahmed, I.; Mir, F.; Ali, S.R.; Zaidi, A.K. Pan-resistant Acinetobacter infection in neonates in Karachi, Pakistan. J. Infect. Dev. Ctries. 2009, 4, 030–037. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Xu, L.; Rysz, M.; Wang, Y.; Zhang, H.; Alvarez, P.J.J. Occurrence and Transport of Tetracycline, Sulfonamide, Quinolone, and Macrolide Antibiotics in the Haihe River Basin, China. Environ. Sci. Technol. 2011, 45, 1827–1833. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, B.; Yu, G. Antibiotic resistance genes in China: Occurrence, risk, and correlation among different parameters. Environ. Sci. Pollut. Res. 2018, 25, 21467–21482. [Google Scholar] [CrossRef] [PubMed]
- Theuretzbacher, U. Accelerating resistance, inadequate antibacterial drug pipelines and international responses. Int. J. Antimicrob. Agents 2012, 39, 295–299. [Google Scholar] [CrossRef]
- Van Schaik, W. The human gut resistome. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Qu, J.; Li, T.; Wirth, S.; Zhang, Y.; Zhao, X.; Li, X. Diet simplification selects for high gut microbial diversity and strong fermenting ability in high-altitude pikas. Appl. Microbiol. Biotechnol. 2018, 102, 6739–6751. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qu, J.; Li, T.; Li, J.; Lin, Q.; Li, X. Pika Population Density Is Associated with the Composition and Diversity of Gut Microbiota. Front. Microbiol. 2016, 7, 758. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, T.; Yao, M.; Li, J.; Zhang, S.; Wirth, S.; Cao, W.; Lin, Q.; Li, X. Pika Gut May Select for Rare but Diverse Environmental Bacteria. Front. Microbiol. 2016, 7, 1269. [Google Scholar] [CrossRef]
- Magoč, T.; Magoč, M.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Thorell, K.; Wurzbacher, C.; Sjöling, Å.; Nilsson, H. Metaxa2 Diversity Tools: Easing microbial community analysis with Metaxa2. Ecol. Informatics 2016, 33, 45–50. [Google Scholar] [CrossRef]
- Bengtsson-Palme, J.; Hartmann, M.; Eriksson, K.; Pal, C.; Thorell, K.; Larsson, D.G.J.; Nilsson, H. metaxa2: Improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol. Ecol. Resour. 2015, 15, 1403–1414. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Hu, X.; Xu, Y.; Xu, W.; Huang, X.; Wen, G.; Yang, K.; Li, Z.; Cao, Y. Persistence and spatial variation of antibiotic resistance genes and bacterial populations change in reared shrimp in South China. Environ. Int. 2018, 119, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-H.; Qiao, M.; Su, J.-Q.; Chen, Z.; Zhou, X.; Zhu, Y.-G. High Throughput Profiling of Antibiotic Resistance Genes in Urban Park Soils with Reclaimed Water Irrigation. Environ. Sci. Technol. 2014, 48, 9079–9085. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.-Y.; Huang, F.-Y.; Zhao, Y.; Li, H.; Su, J.-Q. Increased levels of antibiotic resistance in urban stream of Jiulongjiang River, China. Appl. Microbiol. Biotechnol. 2015, 99, 5697–5707. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2011, 14, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Villafañe, J.H.; Drago, L. What is the site of pain osteoarthritis? A triple gut-brain-joint microbioma axis. Clin. Exp. Rheumatol. 2019, 122, 20–21. [Google Scholar]
- Keohane, D.M.; Woods, T.; O’Connor, P.; Underwood, S.; Cronin, O.; Whiston, R.; O’Sullivan, O.; Cotter, P.; Shanahan, F.; Molloy, M.G. Four men in a boat: Ultra-endurance exercise alters the gut microbiome. J. Sci. Med. Sport 2019, 22, 1059–1064. [Google Scholar] [CrossRef]
- Rettedal, E.A.; Cree, J.M.E.; Adams, S.E.; MacRae, C.; Skidmore, P.M.L.; Cameron-Smith, D.; Gant, N.; Blenkiron, C.; Merry, T.L. Short-term high-intensity interval training exercise does not affect gut bacterial community diversity or composition of lean and overweight men. Exp. Physiol. 2020, 105, 1268–1279. [Google Scholar] [CrossRef]
- Schlomann, B.H.; Parthasarathy, R. Timescales of gut microbiome dynamics. Curr. Opin. Microbiol. 2019, 50, 56–63. [Google Scholar] [CrossRef]
- Mehta, R.S.; Abu-Ali, G.S.; Drew, D.A.; Lloyd-Price, J.; Subramanian, A.; Lochhead, P.; Joshi, A.D.; Ivey, K.L.; Khalili, H.; Brown, G.T.; et al. Stability of the human faecal microbiome in a cohort of adult men. Nat. Microbiol. 2018, 3, 347–355. [Google Scholar] [CrossRef]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The Long-Term Stability of the Human Gut Microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in Fecal Microbiota in Different European Study Populations in Relation to Age, Gender, and Country: A Cross-Sectional Study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, I.; Stegen, J.C.; Maldonado-Gómez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The Gut Microbiota of Rural Papua New Guineans: Composition, Diversity Patterns, and Ecological Processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Blekhman, R.; Goodrich, J.K.; Huang, K.; Sun, Q.; Bukowski, R.; Bell, J.T.; Spector, T.D.; Keinan, A.; Ley, R.E.; Gevers, D.; et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhernakova, A.; Kurilshikov, A.; Bonder, M.J.; Tigchelaar, E.F.; Schirmer, M.; Vatanen, T.; Mujagic, Z.; Vila, A.V.; Falony, G.; Vieira-Silva, S.; et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016, 352, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.J.; Lauber, C.; Costello, E.K.; A Lozupone, C.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting family members share microbiota with one another and with their dogs. eLife 2013, 2, e00458. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking Long-Term Dietary Patterns with Gut Microbial Enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet Drives Convergence in Gut Microbiome Functions Across Mammalian Phylogeny and Within Humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partula, V.; Mondot, S.; Torres, M.J.; Kesse-Guyot, E.; Deschasaux, M.; Assmann, K.; Latino-Martel, P.; Buscail, C.; Julia, C.; Galan, P.; et al. Associations between usual diet and gut microbiota composition: Results from the Milieu Intérieur cross-sectional study. Am. J. Clin. Nutr. 2019, 109, 1472–1483. [Google Scholar] [CrossRef]
- Chen, S.; Cheng, H.; Wyckoff, K.N.; He, Q. Linkages of Firmicutes and Bacteroidetes populations to methanogenic process performance. J. Ind. Microbiol. Biotechnol. 2016, 43, 771–781. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.; Deppenmeier, U. Physiology and central carbon metabolism of the gut bacterium Prevotella copri. Mol. Microbiol. 2018, 109, 528–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taillefer, M.; Arntzen, M.Ø.; Henrissat, B.; Pope, P.; Larsbrink, J. Proteomic Dissection of the Cellulolytic Machineries Used by Soil-DwellingBacteroidetes. mSystems 2018, 3, e00240-18. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, M.; Calusinska, M.; Goux, X.; Rouland-LeFèvre, C.; Untereiner, B.; Ferrer, P.; Gerin, P.A.; Delfosse, P. Carbohydrate Hydrolytic Potential and Redundancy of an Anaerobic Digestion Microbiome Exposed to Acidosis, as Uncovered by Metagenomics. Appl. Environ. Microbiol. 2019, 85, e00895-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, P.O.; Martin, J.C.; Lawley, T.D.; Browne, H.P.; Harris, H.M.B.; Bernalier-Donadille, A.; Duncan, S.H.; O’Toole, P.W.; Scott, K.P.; Flint, H.J. Polysaccharide utilization loci and nutritional specialization in a dominant group of butyrate-producing human colonic Firmicutes. Microb. Genom. 2016, 2, e000043. [Google Scholar] [CrossRef]
- Brown, R.L.; Larkinson, M.L.Y.; Clarke, T.B. Immunological design of commensal communities to treat intestinal infection and inflammation. PLOS Pathog. 2021, 17, e1009191. [Google Scholar] [CrossRef]
- Hollister, E.B.; Gao, C.; Versalovic, J. Compositional and Functional Features of the Gastrointestinal Microbiome and Their Effects on Human Health. Gastroenterology 2014, 146, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Gallegos, R.; Matano, L.M.; Butler, N.L.; Hantman, N.; Kaili, M.; Coyne, M.J.; Comstock, L.E.; Malamy, M.H.; Barquera, B. Genetic and Biochemical Analysis of Anaerobic Respiration inBacteroides fragilisand Its ImportanceIn Vivo. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovatcheva-Datchary, P.; Nilsson, A.; Akrami, R.; Lee, Y.S.; De Vadder, F.; Arora, T.; Hallen, A.; Martens, E.; Björck, I.; Bäckhed, F. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab. 2015, 22, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorvitovskaia, A.; Holmes, S.P.; Huse, S.M. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome 2016, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Koren, O.; Knights, D.; Gonzalez, A.; Waldron, L.; Segata, N.; Knight, R.; Huttenhower, C.; Ley, R.E. A Guide to Enterotypes across the Human Body: Meta-Analysis of Microbial Community Structures in Human Microbiome Datasets. PLoS Comput. Biol. 2013, 9, e1002863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E. Prevotella in the gut: Choose carefully. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.S.; Gupta, S.S.; Nair, G.B.; Mande, S.S. In Silico Analysis of Antibiotic Resistance Genes in the Gut Microflora of Individuals from Diverse Geographies and Age-Groups. PLoS ONE 2013, 8, e83823. [Google Scholar] [CrossRef] [Green Version]
- Gerzova, L.; Videnska, P.; Faldynova, M.; Sedlar, K.; Provaznik, I.; Cizek, A.; Rychlik, I. Characterization of Microbiota Composition and Presence of Selected Antibiotic Resistance Genes in Carriage Water of Ornamental Fish. PLoS ONE 2014, 9, e103865. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Zhang, J.; Chen, H.; Wang, J.; Sun, W.; Zhang, X.; Yang, Y.; Wang, Q.; Ma, J. Effect of temperature on sulfonamide antibiotics degradation, and on antibiotic resistance determinants and hosts in animal manures. Sci. Total Environ. 2017, 607–608, 725–732. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Permanova | Mantel Test | |||||||
|---|---|---|---|---|---|---|---|---|
| Unweighted UniFrac | Weighted UniFrac | Unweighted UniFrac | Weighted UniFrac | |||||
| R2 | p | R2 | p | r | p | r | p | |
| Country | 0.1023 | <0.001 | 0.3120 | <0.001 | 0.658 | <0.001 | 0.583 | <0.001 |
| BMI | 0.0731 | <0.001 | 0.2280 | <0.001 | 0.401 | <0.001 | 0.418 | <0.001 |
| Exercise | 0.0162 | 0.515 | 0.0116 | 0.645 | −0.038 | 0.754 | −0.059 | 0.839 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, T.; Hilal, M.G.; Wang, Y.; Zhou, R.; Yu, Q.; Qu, J.; Li, H. Differences in Gut Microbiome Composition and Antibiotic Resistance Gene Distribution between Chinese and Pakistani University Students from a Common Peer Group. Microorganisms 2021, 9, 1152. https://doi.org/10.3390/microorganisms9061152
Feng T, Hilal MG, Wang Y, Zhou R, Yu Q, Qu J, Li H. Differences in Gut Microbiome Composition and Antibiotic Resistance Gene Distribution between Chinese and Pakistani University Students from a Common Peer Group. Microorganisms. 2021; 9(6):1152. https://doi.org/10.3390/microorganisms9061152
Chicago/Turabian StyleFeng, Tianshu, Mian Gul Hilal, Yijie Wang, Rui Zhou, Qiaoling Yu, Jiapeng Qu, and Huan Li. 2021. "Differences in Gut Microbiome Composition and Antibiotic Resistance Gene Distribution between Chinese and Pakistani University Students from a Common Peer Group" Microorganisms 9, no. 6: 1152. https://doi.org/10.3390/microorganisms9061152