
Visualization of Gene Reciprocity among Lactic Acid Bacteria in Yogurt by RNase H-Assisted Rolling Circle Amplification-Fluorescence In Situ Hybridization

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Padlock Probe and Detection Probe
2.2. Real-Time RHa-RCA
2.3. Yogurt Fermentation and LAB Collection
2.4. Cell Fixation and Permeabilization
2.5. In Situ mRNA Detection Using RHa-RCA-FISH
2.6. Imaging and Analysis
3. Results
3.1. Collection of LAB Cells from Fermented Yogurt
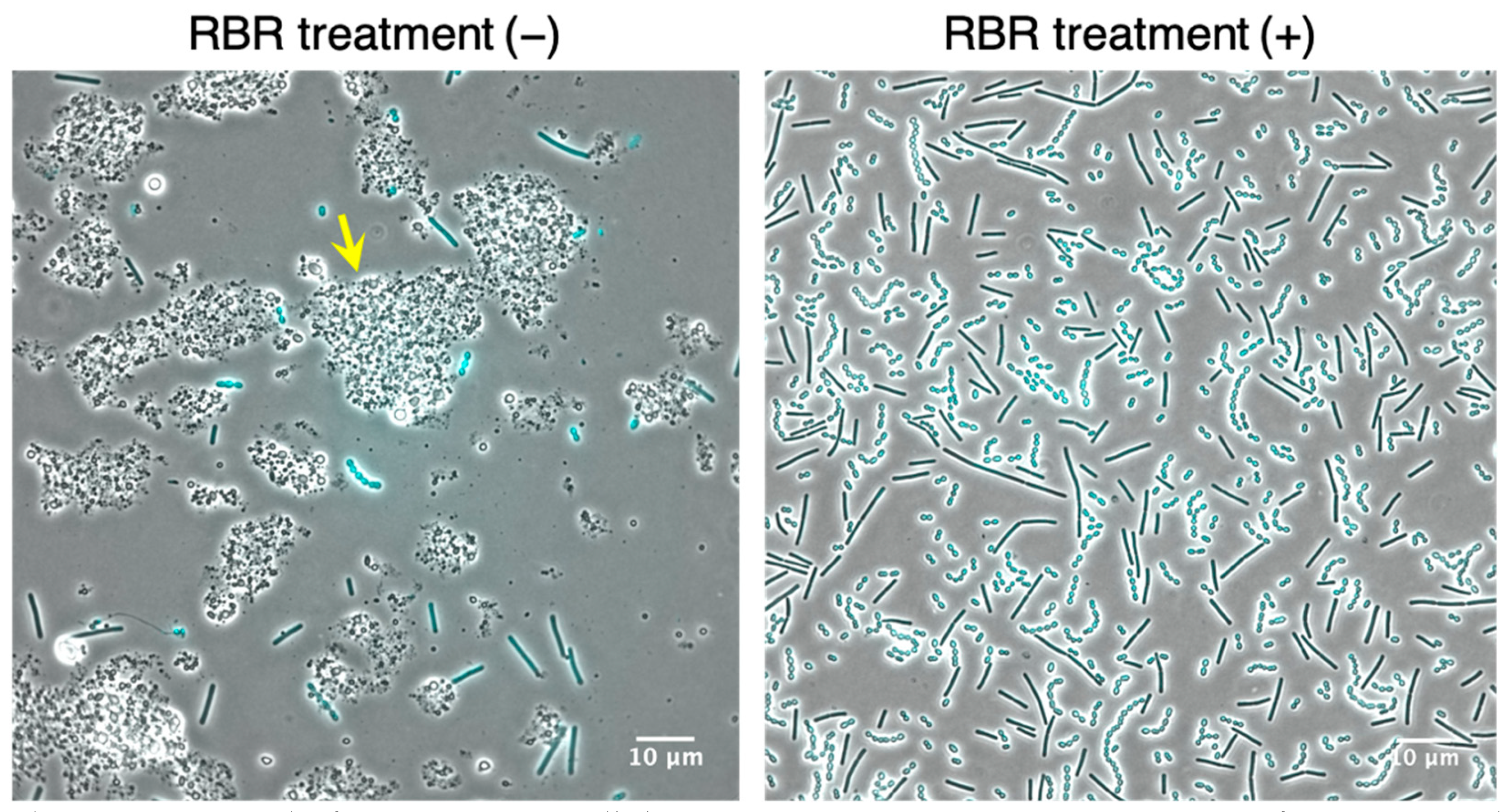
3.2. Establishment of the Amount of Lysozyme Sufficient for LAB Cells Permeabilization
3.3. Monitoring of the Temporal Transition of pflA Expression in S. thermophilus Cells by RHa-RCA-FISH
3.4. Selection of PLP for Detecting ldhD1 mRNA of L. bulgaricus
3.5. Monitoring of the Temporal Transition of ldhD1 Expression in L. bulgaricus Cells by RHa-RCA-FISH
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kruse, S.; Goris, T.; Westermann, M.; Adrian, L.; Diekert, G. Hydrogen production by Sulfurospirillum species enables syntrophic interactions of Epsilonproteobacteria. Nat. Commun. 2018, 9, 4872. [Google Scholar] [CrossRef] [Green Version]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic Identification and In Situ Detection of Individual Microbial Cells without Cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Jansson, J.K.; Neufeld, J.D.; Moran, M.A.; Gilbert, J.A. Omics for understanding microbial functional dynamics. Environ. Microbiol. 2012, 14, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pester, M.; Bittner, N.; Deevong, P.; Wagner, M.; Loy, A. A ‘rare biosphere’ microorganism contributes to sulfate reduction in a peatland. ISME J. 2010, 4, 1591–1602. [Google Scholar] [CrossRef]
- Bodelier, P.L.E.; Meima-Franke, M.; Hordijk, C.A.; Steenbergh, A.K.; Hefting, M.M.; Bodrossy, L.; von Bergen, M.; Seifert, J. Microbial minorities modulate methane consumption through niche partitioning. ISME J. 2013, 7, 2214–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, C.E.; Strachan, B.J.; Hanson, N.W.; Hahn, A.S.; Hall, E.R.; Rabinowitz, B.; Mavinic, D.S.; Ramey, W.D.; Hallam, S.J. Rare taxa have potential to make metabolic contributions in enhanced biological phosphorus removal ecosystems. Environ. Microbiol. 2015, 17, 4979–4993. [Google Scholar] [CrossRef]
- Sato, Y.; Hori, T.; Koike, H.; Navarro, R.R.; Ogata, A.; Habe, H. Transcriptome analysis of activated sludge microbiomes reveals an unexpected role of minority nitrifiers in carbon metabolism. Comm. Biol. 2019, 2, 179. [Google Scholar] [CrossRef] [Green Version]
- Raj, A.; Peskin, C.S.; Tranchina, D.; Vargas, D.Y.; Tyagi, S. Stochastic mRNA Synthesis in Mammalian Cells. PLoS Biol. 2006, 4, e309. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [Green Version]
- So, L.H.; Ghosh, A.; Zong, C.; Sepúlveda, L.A.; Segev, R.; Golding, I. General properties of transcriptional time series in Escherichia coli. Nat. Genet. 2011, 43, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Skinner, S.O.; Sepúlveda, L.A.; Xu, H.; Golding, I. Measuring mRNA copy number in individual Escherichia coli cells using single-molecule fluorescent in situ hybridization. Nat. Protoc. 2013, 8, 1100–1113. [Google Scholar] [CrossRef]
- Badstöber, J.; Gachon, C.M.M.; Ludwig-Müller, J.; Sandbichler, A.M.; Neuhauser, S. Demystifying biotrophs: FISHing for mRNAs to decipher plant and algal pathogen–host interaction at the single cell level. Sci. Rep. 2020, 10, 14269. [Google Scholar] [CrossRef]
- Takahashi, H.; Ohkawachi, M.; Horio, K.; Kobori, T.; Aki, T.; Matsumura, Y.; Nakashimada, Y.; Okamura, Y. RNase H-assisted RNA-primed rolling circle amplification for targeted RNA sequence detection. Sci. Rep. 2018, 8, 7770. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Horio, K.; Kato, S.; Kobori, T.; Watanabe, K.; Aki, T.; Nakashimada, Y.; Okamura, Y. Direct detection of mRNA expression in microbial cells by fluorescence in situ hybridization using RNase H-assisted rolling circle amplification. Sci. Rep. 2020, 10, 9588. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, I.; Kato, S.; Kitada, T.; Yano, N.; Morichi, T. Growth of Lactobacillus bulgaricus in Milk. 1. Cell Elongation and the Role of Formic Acid in Boiled Milk. J. Dairy Sci. 1986, 69, 311–320. [Google Scholar] [CrossRef]
- Sieuwerts, S.; Molenaar, D.; van Hijum, S.A.F.T.; Beerthuyzen, M.; Stevens, M.J.A.; Janssen, P.W.M.; Ingham, C.J.; de Bok, F.A.M.; de Vos, W.M.; van Hylckama Vlieg, J.E.T. Mixed-Culture Transcriptome Analysis Reveals the Molecular Basis of Mixed-Culture Growth in Streptococcus thermophilus and Lactobacillus bulgaricus. Appl. Environ. Microbiol. 2010, 76, 7775–7784. [Google Scholar] [CrossRef] [Green Version]
- de Bok, F.A.M.; Janssen, P.W.M.; Bayjanov, J.R.; Sieuwerts, S.; Lommen, A.; van Hylckama Vlieg, J.E.T.; Molenaar, D. Volatile Compound Fingerprinting of Mixed-Culture Fermentations. Appl. Environ. Microbiol. 2011, 77, 6233–6239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceruso, M.; Liu, Y.; Gunther, N.W., IV; Pepe, T.; Anastasio, A.; Qi, P.X.; Tomasula, P.M.; Renye, J.A., Jr. Anti-listerial activity of thermophilin 110 and pediocin in fermented milk and whey. Food Control 2021, 125, 107941. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, E.; Shi, T.; Ye, L.; Konno, T.; Oda, M.; Ji, Z.S. Strand-specific RNA-seq analysis of the Lactobacillus delbrueckii subsp. bulgaricus transcriptome. Mol. BioSyst. 2016, 12, 508–519. [Google Scholar] [CrossRef]
- Li, H.; Yang, C.; Chen, C.; Ren, F.; Li, Y.; Mu, Z.; Wang, P. The Use of Trisodium Citrate to Improve the Textural Properties of Acid-Induced, Transglutaminase-Treated Micellar Casein Gels. Molecules 2018, 23, 1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, H.; Kim, J.D. Simple and Rapid Method for Separating Lactic Acid Bacteria from Commercially Prepared Yogurt. Anal. Sci. 2019, 35, 1065–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Satoh, T.; Kanahara, H.; Kubota, Y.; Hirose, T.; Yamazaki, H.; Yamamoto, K.; Okamura, Y.; Suzuki, T.; Kobori, T. Development of a bench-top extracleanroom for DNA amplification. BioTechniques 2016, 61, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Saulnier, D.M.A.; Molenaar, D.; de Vos, W.M.; Gibson, G.R.; Kolida, S. Identification of Prebiotic Fructooligosaccharide Metabolism in Lactobacillus plantarum WCFS1 through Microarrays. Appl. Environ. Microbiol. 2007, 73, 1753–1765. [Google Scholar] [CrossRef] [Green Version]
- Gunasekera, T.S.; Attfield, P.V.; Veal, D.A. A Flow Cytometry Method for Rapid Detection and Enumeration of Total Bacteria in Milk. Appl. Environ. Microbiol. 2000, 66, 1228–1232. [Google Scholar] [CrossRef] [Green Version]
- Gunasekera, T.S.; Sørensen, A.; Attfield, P.V.; Sørensen, S.J.; Veal, D.A. Inducible Gene Expression by Nonculturable Bacteria in Milk after Pasteurization. Appl. Environ. Microbiol. 2002, 68, 1988–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeianov, V.V.; Wechter, P.; Broadbent, J.R.; Hughes, J.E.; Rodríguez, B.T.; Christensen, T.K.; Ardö, Y.; Steele, J.L. Comparative High-Density Microarray Analysis of Gene Expression during Growth of Lactobacillus helveticus in Milk versus Rich Culture Medium. Appl. Environ. Microbiol. 2007, 73, 2661–2672. [Google Scholar] [CrossRef] [Green Version]
- Macklaim, J.M.; Fernandes, A.D.; Di Bella, J.M.; Hammond, J.A.; Reid, G.; Gloor, G.B. Comparative meta-RNA-seq of the vaginal microbiota and differential expression by Lactobacillus iners in health and dysbiosis. Microbiome 2013, 1, 12. [Google Scholar] [CrossRef] [Green Version]
- Bisanz, J.E.; Macklaim, J.M.; Gloor, G.B.; Reid, G. Bacterial metatranscriptome analysis of a probiotic yogurt using an RNA-Seq approach. Int. Dairy J. 2014, 39, 284–292. [Google Scholar] [CrossRef]
- Schneider, N.; Meier, M. Efficient in situ detection of mRNAs using the Chlorella virus DNA ligase for padlock probe ligation. RNA 2017, 23, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Rouskin, S.; Zubradt, M.; Washietl, S.; Kellis, M.; Weissman, J.S. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 2014, 505, 701–705. [Google Scholar] [CrossRef] [Green Version]
- Engl, C. Noise in bacterial gene expression. Biochem. Soc. Trans. 2019, 47, 209–217. [Google Scholar] [CrossRef]
- Grieb, A.; Bowers, R.M.; Oggerin, M.; Goudeau, D.; Lee, J.; Malmstrom, R.R.; Woyke, T.; Fuchs, B.M. A pipeline for targeted metagenomics of environmental bacteria. Microbiome 2020, 8, 21. [Google Scholar] [CrossRef]
- Dou, D.; Hernández-Neuta, I.; Wang, H.; Östbye, H.; Qian, X.; Thiele, S.; Resa-Infante, P.; Kouassi, N.M.; Sender, V.; Hentrich, K.; et al. Analysis of IAV Replication and Co-infection Dynamics by a Versatile RNA Viral Genome Labeling Method. Cell Rep. 2017, 20, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudent, E.; Lepidi, H.; Angelakis, E.; Raoult, D. Fluorescence In Situ Hybridization (FISH) and Peptide Nucleic Acid Probe-Based FISH for Diagnosis of Q Fever Endocarditis and Vascular Infections. J. Clin. Microbiol. 2018, 56, e00542-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukumbuzya, M.; Schmid, M.; Pjevac, P.; Daims, H. A Multicolor Fluorescence in situ Hybridization Approach Using an Extended Set of Fluorophores to Visualize Microorganisms. Front Microbiol. 2019, 10, 1383. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Kamagata, Y.; Nakamura, K.; Ohashi, A.; Harada, H. Fluorescence In Situ Hybridization Using 16S rRNA-Targeted Oligonucleotides Reveals Localization of Methanogens and Selected Uncultured Bacteria in Mesophilic and Thermophilic Sludge Granules. Appl. Environ. Microbiol. 1999, 65, 1280–1288. [Google Scholar] [CrossRef] [Green Version]
- Dige, I.; Nilsson, H.; Kilian, M.; Nyvad, B. In situ identification of streptococci and other bacteria in initial dental biofilm by confocal laser scanning microscopy and fluorescence in situ hybridization. Eur. J. Oral Sci. 2007, 115, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, M.; Luvisi, A.; Meyer, J.B.; Sabella, E.; De Bellis, L.; Cruz, A.C.; Ampatzidis, Y.; Cherubini, P. Specific Fluorescence in Situ Hybridization (FISH) Test to Highlight Colonization of Xylem Vessels by Xylella fastidiosa in Naturally Infected Olive Trees (Olea europaea L.). Front. Plant Sci. 2018, 9, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, N.; Pollock, F.J.; Willis, B.L.; Ainsworth, T.; Mano, N.; Bourne, D.G. In situ visualization of bacterial populations in coral tissues: Pitfalls and solutions. PeerJ 2016, 4, e2424. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horio, K.; Takahashi, H.; Kobori, T.; Watanabe, K.; Aki, T.; Nakashimada, Y.; Okamura, Y. Visualization of Gene Reciprocity among Lactic Acid Bacteria in Yogurt by RNase H-Assisted Rolling Circle Amplification-Fluorescence In Situ Hybridization. Microorganisms 2021, 9, 1208. https://doi.org/10.3390/microorganisms9061208
Horio K, Takahashi H, Kobori T, Watanabe K, Aki T, Nakashimada Y, Okamura Y. Visualization of Gene Reciprocity among Lactic Acid Bacteria in Yogurt by RNase H-Assisted Rolling Circle Amplification-Fluorescence In Situ Hybridization. Microorganisms. 2021; 9(6):1208. https://doi.org/10.3390/microorganisms9061208
Chicago/Turabian StyleHorio, Kyohei, Hirokazu Takahashi, Toshiro Kobori, Kenshi Watanabe, Tsunehiro Aki, Yutaka Nakashimada, and Yoshiko Okamura. 2021. "Visualization of Gene Reciprocity among Lactic Acid Bacteria in Yogurt by RNase H-Assisted Rolling Circle Amplification-Fluorescence In Situ Hybridization" Microorganisms 9, no. 6: 1208. https://doi.org/10.3390/microorganisms9061208
APA StyleHorio, K., Takahashi, H., Kobori, T., Watanabe, K., Aki, T., Nakashimada, Y., & Okamura, Y. (2021). Visualization of Gene Reciprocity among Lactic Acid Bacteria in Yogurt by RNase H-Assisted Rolling Circle Amplification-Fluorescence In Situ Hybridization. Microorganisms, 9(6), 1208. https://doi.org/10.3390/microorganisms9061208