
Salivary Microbiome Diversity in Kuwaiti Adolescents with Varied Body Mass Index—A Pilot Study

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction and 16S rRNA Gene Sequencing
2.3. Bioinformatics Analyses
3. Results
3.1. Sample Characteristics
3.2. Amplicon Sequencing Data Analyses
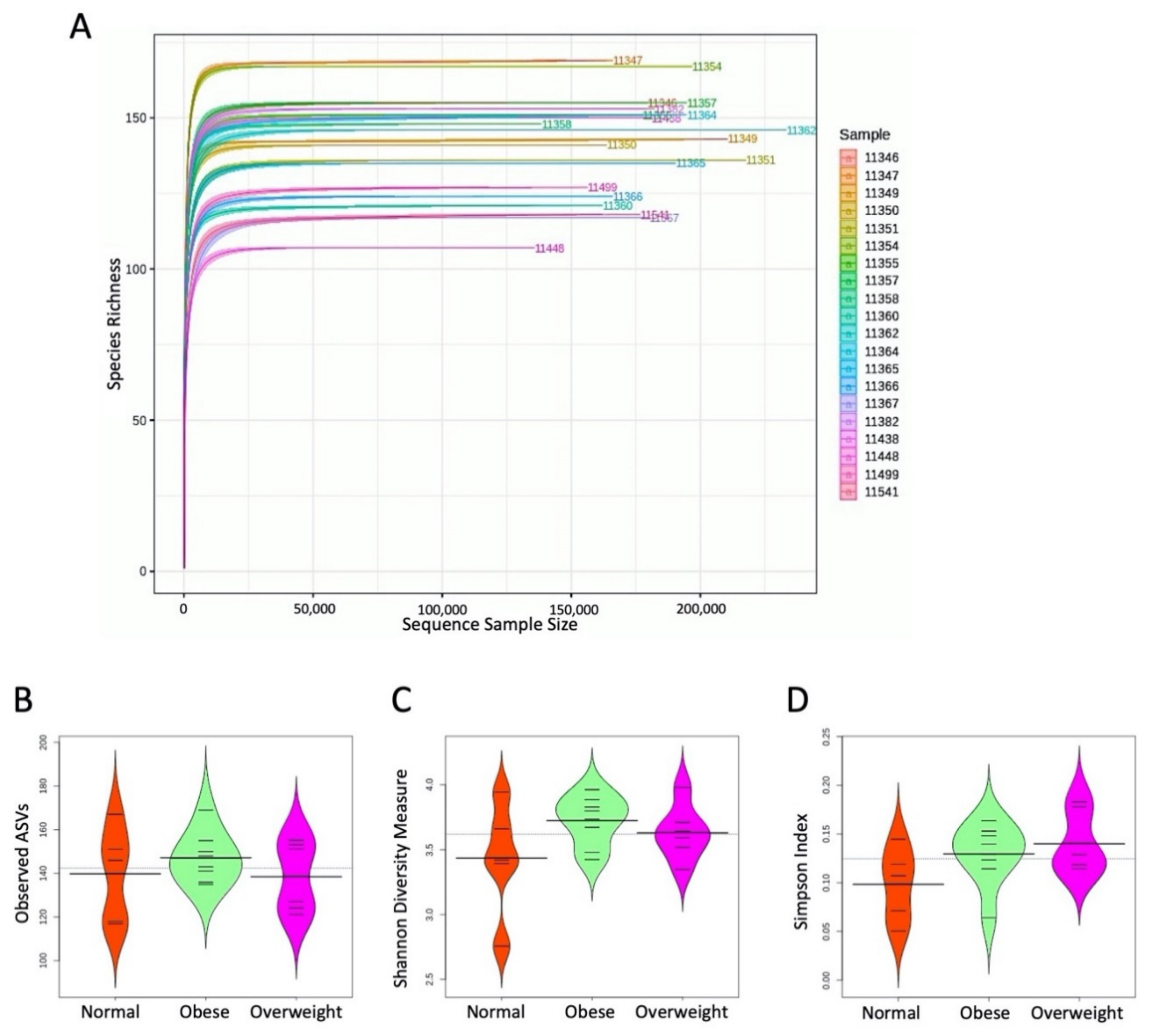
3.3. Alpha Diversity
3.4. Beta Diversity
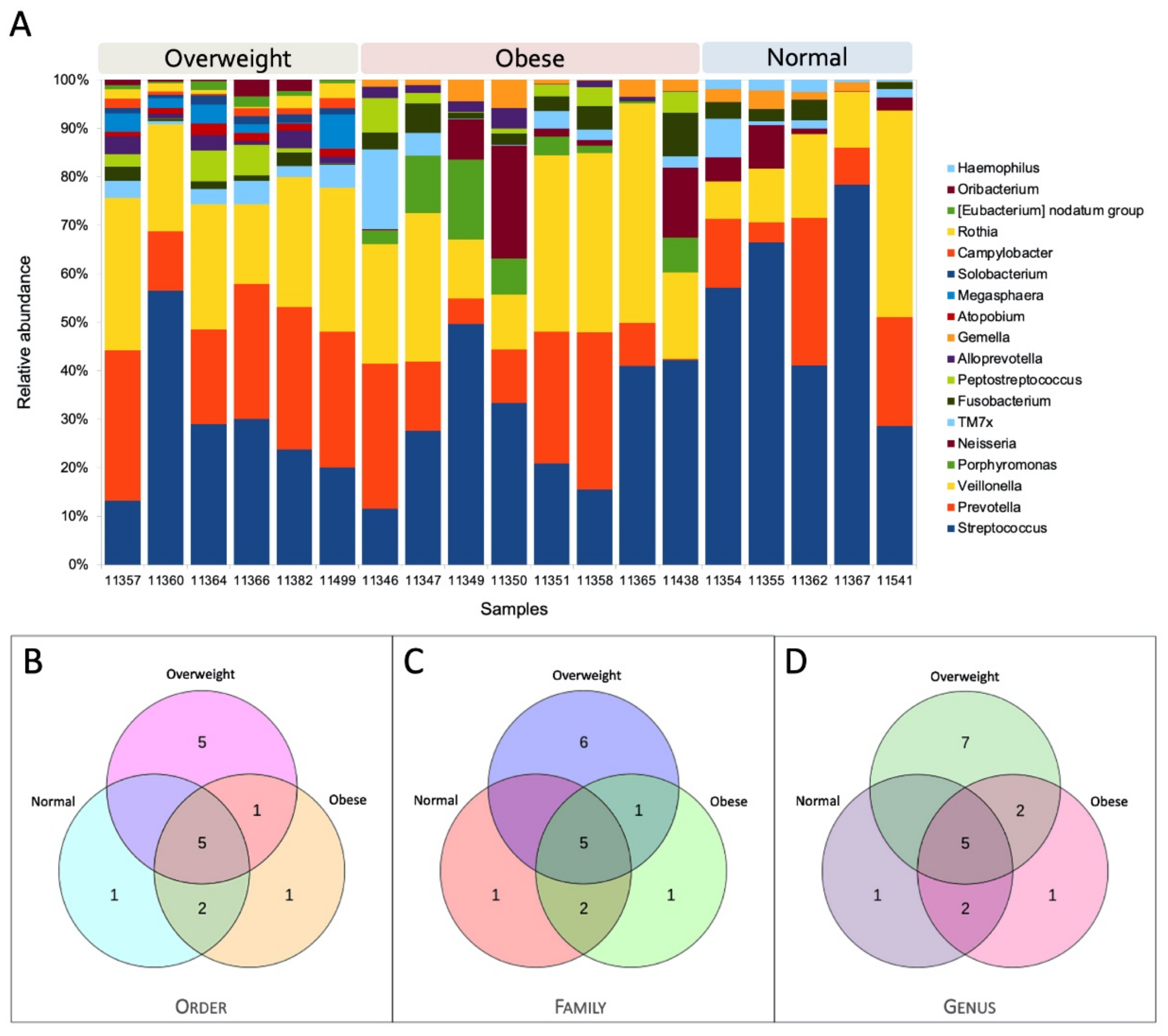
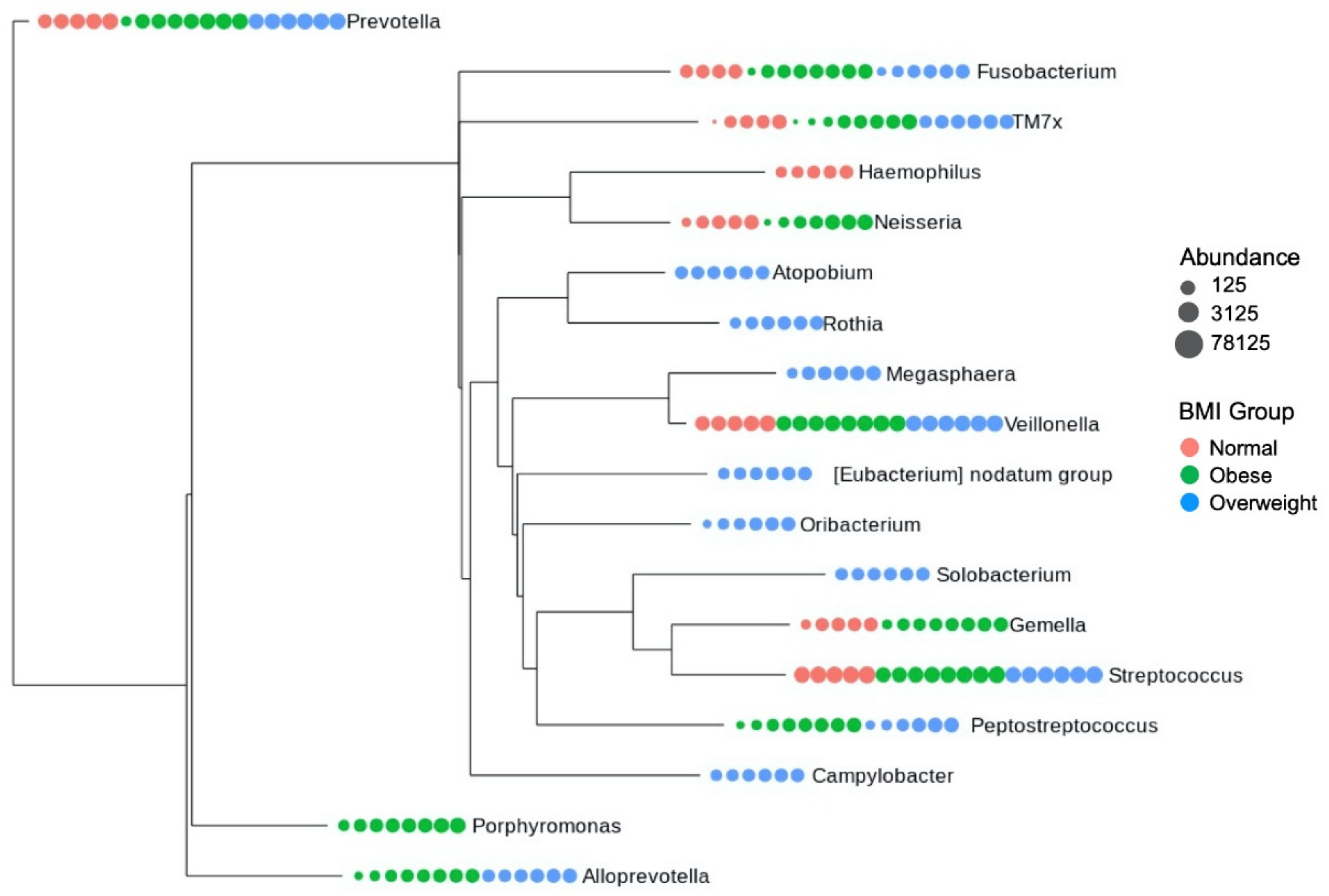
3.5. Core Microbiome Structure
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization Noncommunicable Diseases Country Profiles 2018; World Health Organization: Geneva, Switzerland, 2018.
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut Microbiota, Lipopolysaccharides, and Innate Immunity in the Pathogenesis of Obesity and Cardiovascular Risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, J.M. The Immune Response to Prevotella Bacteria in Chronic Inflammatory Disease. Immunology 2017, 151, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of Human Gut Microbiome Correlates with Metabolic Markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Yamazaki, K. Can Oral Bacteria Affect the Microbiome of the Gut? J. Oral Microbiol. 2019, 11, 1586422. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.S.; Hayward, M.R.; Coelho, L.P.; Li, S.S.; Costea, P.I.; Voigt, A.Y.; Wirbel, J.; Maistrenko, O.M.; Alves, R.J.; Bergsten, E.; et al. Extensive Transmission of Microbes along the Gastrointestinal Tract. Elife 2019, 8. [Google Scholar] [CrossRef]
- Human Microbiome Project Consortium Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [CrossRef] [Green Version]
- Belstrøm, D. The Salivary Microbiota in Health and Disease. J. Oral Microbiol. 2020, 12, 1723975. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chi, X.; Zhang, Q.; Chen, F.; Deng, X. Characterization of the Salivary Microbiome in People with Obesity. PeerJ 2018, 6, e4458. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.; Hoffmann, T.; Fischer, S.; Bornstein, S.; Gräßler, J.; Noack, B. Obesity Alters Composition and Diversity of the Oral Microbiota in Patients with Type 2 Diabetes Mellitus Independently of Glycemic Control. PLoS ONE 2018, 13, e0204724. [Google Scholar] [CrossRef]
- Mervish, N.A.; Hu, J.; Hagan, L.A.; Arora, M.; Frau, C.; Choi, J.; Attaie, A.; Ahmed, M.; Teitelbaum, S.L.; Wolff, M.S. Associations of the Oral Microbiota with Obesity and Menarche in Inner City Girls. J Child. Obes 2019, 4. [Google Scholar] [CrossRef]
- De Andrade, P.A.M.; Giovani, P.A.; Araujo, D.S.; de Souza, A.J.; Pedroni-Pereira, A.; Kantovitz, K.R.; Andreote, F.D.; Castelo, P.M.; Nociti, F.H., Jr. Shifts in the Bacterial Community of Saliva Give Insights on the Relationship between Obesity and Oral Microbiota in Adolescents. Arch. Microbiol. 2020, 202, 1085–1095. [Google Scholar] [CrossRef]
- Araujo, D.S.; Klein, M.I.; Scudine, K.G.d.O.; de Sales Leite, L.; Parisotto, T.M.; Ferreira, C.M.; Fonseca, F.L.A.; Perez, M.M.; Castelo, P.M. Salivary Microbiological and Gingival Health Status Evaluation of Adolescents With Overweight and Obesity: A Cluster Analysis. Front. Pediatr. 2020, 8, 429. [Google Scholar] [CrossRef]
- Murugesan, S.; Al Ahmad, S.F.; Singh, P.; Saadaoui, M.; Kumar, M.; Al Khodor, S. Profiling the Salivary Microbiome of the Qatari Population. J. Transl. Med. 2020, 18, 127. [Google Scholar] [CrossRef] [PubMed]
- Alqaderi, H.; Al-Ozairi, E.; Bin-Hasan, S.; Tavares, M.; Goodson, J.M.; Alsumait, A.; Abu-Farha, M.; Abubaker, J.; Devarajan, S.; Almuhana, N.; et al. Mediation Effect of C-Reactive Protein in the Relationship between Abdominal Obesity and Intermediate Hyperglycemia in Kuwaiti Adolescents. Biomark. Med. 2020, 14, 1427–1437. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, V.; Song, X.; Asquith, M.; Karstens, L. Performance of Microbiome Sequence Inference Methods in Environments with Varying Biomass. mSystems 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Murali, A.; Bhargava, A.; Wright, E.S. IDTAXA: A Novel Approach for Accurate Taxonomic Classification of Microbiome Sequences. Microbiome 2018, 6, 140. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakodi, M.P. Effect of Amplicon Sequencing Depth in Environmental Microbiome Research. Curr. Microbiol. 2021, 78, 1026–1033. [Google Scholar] [CrossRef]
- Xiao, J.; Fiscella, K.A.; Gill, S.R. Oral Microbiome: Possible Harbinger for Children’s Health. Int. J. Oral Sci. 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.G.; Carey, H.V.; Domazet-Lošo, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a Bacterial World, a New Imperative for the Life Sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, N.A. A Common Origin for Immunity and Digestion. Front. Immunol. 2015, 6, 72. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th edition) 1. Autophagy 2021, 17, 1–382. [Google Scholar] [PubMed]
- Dewi, L.; Rosidi, A.; Noer, E.R.; Ayuningtyas, A. Prospect for Type 2 Diabetes Mellitus in the Combination of Exercise and Synbiotic: A Perspective. Curr. Diabetes Rev. 2021. [Google Scholar] [CrossRef]
- Troisi, J.; Venutolo, G.; Pujolassos Tanyà, M.; Delli Carri, M.; Landolfi, A.; Fasano, A. COVID-19 and the Gastrointestinal Tract: Source of Infection or Merely a Target of the Inflammatory Process Following SARS-CoV-2 Infection? World J. Gastroenterol. 2021, 27, 1406–1418. [Google Scholar] [CrossRef]
- Raju, S.C.; Lagström, S.; Ellonen, P.; de Vos, W.M.; Eriksson, J.G.; Weiderpass, E.; Rounge, T.B. Gender-Specific Associations Between Saliva Microbiota and Body Size. Front. Microbiol. 2019, 10, 767. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A Core Gut Microbiome in Obese and Lean Twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Sze, M.A.; Schloss, P.D. Looking for a Signal in the Noise: Revisiting Obesity and the Microbiome. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, B.A.; Shapiro, J.A.; Church, T.R.; Miller, G.; Trinh-Shevrin, C.; Yuen, E.; Friedlander, C.; Hayes, R.B.; Ahn, J. A Taxonomic Signature of Obesity in a Large Study of American Adults. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanislawski, M.A.; Dabelea, D.; Lange, L.A.; Wagner, B.D.; Lozupone, C.A. Gut Microbiota Phenotypes of Obesity. NPJ Biofilms Microbiomes 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Alshihri, A.A.; Rogers, H.J.; Alqahtani, M.A.; Aldossary, M.S. Association between Dental Caries and Obesity in Children and Young People: A Narrative Review. Int. J. Dent. 2019, 2019, 9105759. [Google Scholar] [CrossRef]
- Aas, J.A.; Paster, B.J.; Stokes, L.N.; Olsen, I.; Dewhirst, F.E. Defining the Normal Bacterial Flora of the Oral Cavity. J. Clin. Microbiol. 2005, 43, 5721–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Dewhirst, F. Human Oral Microbiome Database (HOMD). Encycl. Metagenom. 2015, 271–291. [Google Scholar]
- Man, S.M. The Clinical Importance of Emerging Campylobacter Species. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 669–685. [Google Scholar] [CrossRef]
- Zeigler, C.C.; Persson, G.R.; Wondimu, B.; Marcus, C.; Sobko, T.; Modéer, T. Microbiota in the Oral Subgingival Biofilm Is Associated with Obesity in Adolescence. Obesity 2012, 20, 157–164. [Google Scholar] [CrossRef]
- Mulhall, H.; Huck, O.; Amar, S. Porphyromonas Gingivalis, a Long-Range Pathogen: Systemic Impact and Therapeutic Implications. Microorganisms 2020, 8. [Google Scholar] [CrossRef]
- Stahringer, S.S.; Clemente, J.C.; Corley, R.P.; Hewitt, J.; Knights, D.; Walters, W.A.; Knight, R.; Krauter, K.S. Nurture Trumps Nature in a Longitudinal Survey of Salivary Bacterial Communities in Twins from Early Adolescence to Early Adulthood. Genome Res. 2012, 22, 2146–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.R.; Yu, W.-H.; Lakshmanan, A.; Wade, W.G. The Human Oral Microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [Green Version]
- Koliada, A.; Syzenko, G.; Moseiko, V.; Budovska, L.; Puchkov, K.; Perederiy, V.; Gavalko, Y.; Dorofeyev, A.; Romanenko, M.; Tkach, S.; et al. Association between Body Mass Index and Firmicutes/Bacteroidetes Ratio in an Adult Ukrainian Population. BMC Microbiol. 2017, 17, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohail, M.U.; Elrayess, M.A.; Al Thani, A.A.; Al-Asmakh, M.; Yassine, H.M. Profiling the Oral Microbiome and Plasma Biochemistry of Obese Hyperglycemic Subjects in Qatar. Microorganisms 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Patil, D.P.; Dhotre, D.P.; Chavan, S.G.; Sultan, A.; Jain, D.S.; Lanjekar, V.B.; Gangawani, J.; Shah, P.S.; Todkar, J.S.; Shah, S.; et al. Molecular Analysis of Gut Microbiota in Obesity among Indian Individuals. J. Biosci. 2012, 37, 647–657. [Google Scholar] [CrossRef]
- Tims, S.; Derom, C.; Jonkers, D.M.; Vlietinck, R.; Saris, W.H.; Kleerebezem, M.; de Vos, W.M.; Zoetendal, E.G. Microbiota Conservation and BMI Signatures in Adult Monozygotic Twins. ISME J. 2013, 7, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12. [Google Scholar] [CrossRef]
- Liu, B.; Faller, L.L.; Klitgord, N.; Mazumdar, V.; Ghodsi, M.; Sommer, D.D.; Gibbons, T.R.; Treangen, T.J.; Chang, Y.-C.; Li, S.; et al. Deep Sequencing of the Oral Microbiome Reveals Signatures of Periodontal Disease. PLoS ONE 2012, 7, e37919. [Google Scholar] [CrossRef] [Green Version]
- D’Angiolella, G.; Tozzo, P.; Gino, S.; Caenazzo, L. Trick or Treating in Forensics—The Challenge of the Saliva Microbiome: A Narrative Review. Microorganisms 2020, 8, 1501. [Google Scholar] [CrossRef]
- Nasidze, I.; Li, J.; Quinque, D.; Tang, K.; Stoneking, M. Global Diversity in the Human Salivary Microbiome. Genome Res. 2009, 19, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Quinque, D.; Horz, H.-P.; Li, M.; Rzhetskaya, M.; Raff, J.A.; Hayes, M.G.; Stoneking, M. Comparative Analysis of the Human Saliva Microbiome from Different Climate Zones: Alaska, Germany, and Africa. BMC Microbiol. 2014, 14, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylro, V.S.; Roesch, L.F.W.; Ortega, J.M.; do Amaral, A.M.; Tótola, M.R.; Hirsch, P.R.; Rosado, A.S.; Góes-Neto, A.; da Costa da Silva, A.L.; Rosa, C.A.; et al. Brazilian Microbiome Project: Revealing the Unexplored Microbial Diversity—Challenges and Prospects. Microb. Ecol. 2014, 67, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozga, A.T.; Sankaranarayanan, K.; Tito, R.Y.; Obregon-Tito, A.J.; Foster, M.W.; Tallbull, G.; Spicer, P.; Warinner, C.G.; Lewis, C.M., Jr. Oral Microbiome Diversity among Cheyenne and Arapaho Individuals from Oklahoma. Am. J. Phys. Anthropol. 2016, 161, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, A.W.; Priya, S.; Blekhman, R.; Bordenstein, S.R. Gut Microbiota Diversity across Ethnicities in the United States. PLoS Biol. 2018, 16, e2006842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, B.; Selvaraju, V.; Chen, J.; Ayine, P.; Yang, L.; Ramesh Babu, J.; Geetha, T.; Taneja, V. Ethnic Variability Associating Gut and Oral Microbiome with Obesity in Children. Gut Microbes 2021, 13, 1–15. [Google Scholar] [CrossRef]
- Eaaswarkhanth, M.; Pathak, A.K.; Ongaro, L.; Montinaro, F.; Hebbar, P.; Alsmadi, O.; Metspalu, M.; Al-Mulla, F.; Thanaraj, T.A. Unraveling a Fine-Scale High Genetic Heterogeneity and Recent Continental Connections of an Arabian Peninsula Population. Eur. J. Hum. Genet. 2021. [Google Scholar] [CrossRef]
- Eaaswarkhanth, M.; Dos Santos, A.L.C.; Gokcumen, O.; Al-Mulla, F.; Thanaraj, T.A. Genome-Wide Selection Scan in an Arabian Peninsula Population Identifies a TNKS Haplotype Linked to Metabolic Traits and Hypertension. Genome Biol. Evol. 2020, 12, 77–87. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqaderi, H.; Ramakodi, M.P.; Nizam, R.; Jacob, S.; Devarajan, S.; Eaaswarkhanth, M.; Al-Mulla, F. Salivary Microbiome Diversity in Kuwaiti Adolescents with Varied Body Mass Index—A Pilot Study. Microorganisms 2021, 9, 1222. https://doi.org/10.3390/microorganisms9061222
Alqaderi H, Ramakodi MP, Nizam R, Jacob S, Devarajan S, Eaaswarkhanth M, Al-Mulla F. Salivary Microbiome Diversity in Kuwaiti Adolescents with Varied Body Mass Index—A Pilot Study. Microorganisms. 2021; 9(6):1222. https://doi.org/10.3390/microorganisms9061222
Chicago/Turabian StyleAlqaderi, Hend, Meganathan P. Ramakodi, Rasheeba Nizam, Sindhu Jacob, Sriraman Devarajan, Muthukrishnan Eaaswarkhanth, and Fahd Al-Mulla. 2021. "Salivary Microbiome Diversity in Kuwaiti Adolescents with Varied Body Mass Index—A Pilot Study" Microorganisms 9, no. 6: 1222. https://doi.org/10.3390/microorganisms9061222
APA StyleAlqaderi, H., Ramakodi, M. P., Nizam, R., Jacob, S., Devarajan, S., Eaaswarkhanth, M., & Al-Mulla, F. (2021). Salivary Microbiome Diversity in Kuwaiti Adolescents with Varied Body Mass Index—A Pilot Study. Microorganisms, 9(6), 1222. https://doi.org/10.3390/microorganisms9061222