
Structural and Functional Impacts of Microbiota on Pyropia yezoensis and Surrounding Seawater in Cultivation Farms along Coastal Areas of the Yellow Sea

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites and Sample Collection
2.2. DNA Extraction
2.3. 16.S rRNA Gene Amplification
2.4. Library Preparation and Sequencing
2.5. Data Processing Based on Bioinformatics and Statistical Analysis
3. Results
3.1. Physico-Chemical Properties of Seawater
3.2. Bacterial Community Diversities
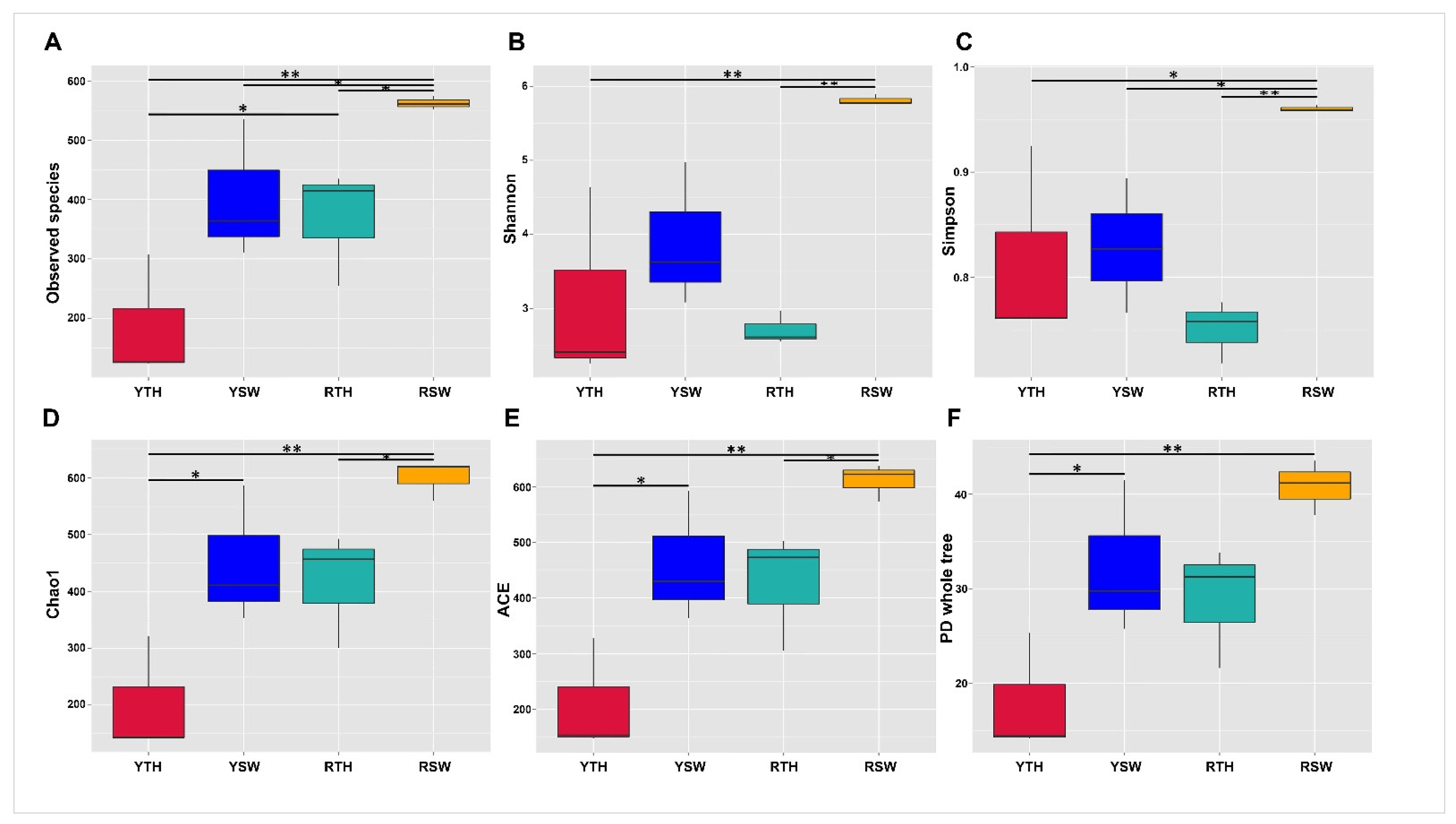
3.2.1. Alpha Diversity
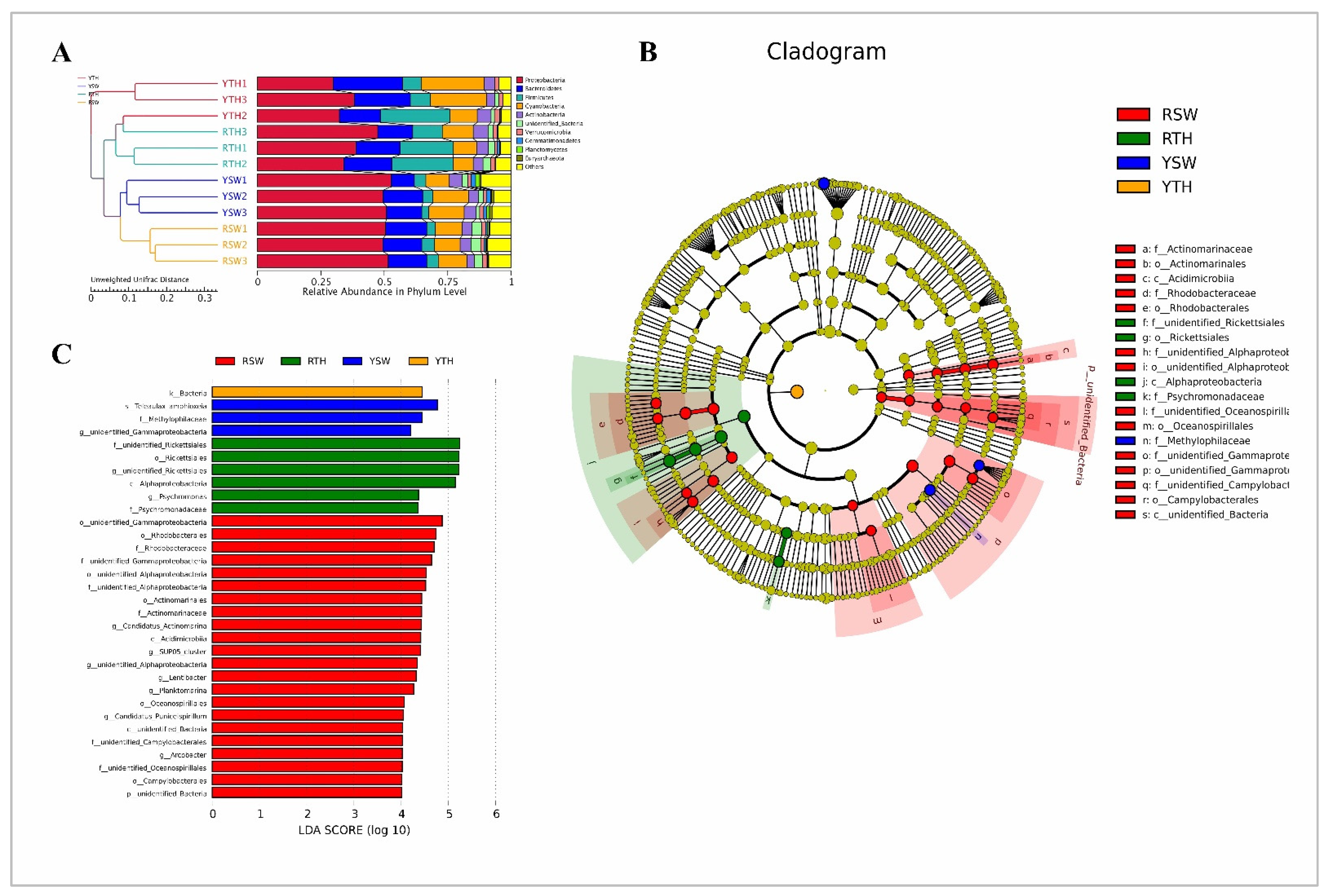
3.2.2. Beta Diversity
3.2.3. Linear Discriminant Analysis Effect Size (LEfSe)
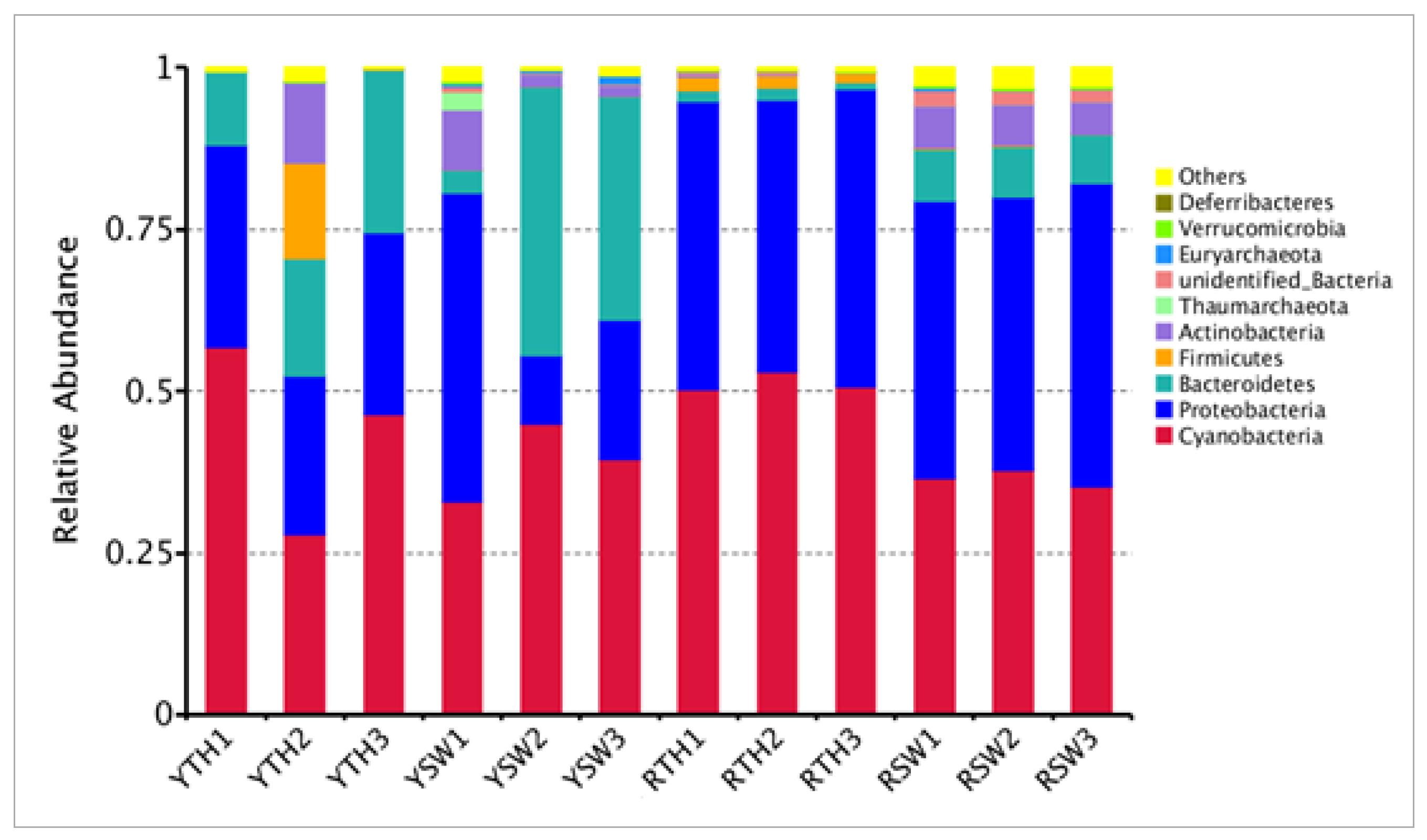
3.3. Composition of Bacterial Community
3.4. PICRUSt Analysis on Metagenome Functions Prediction
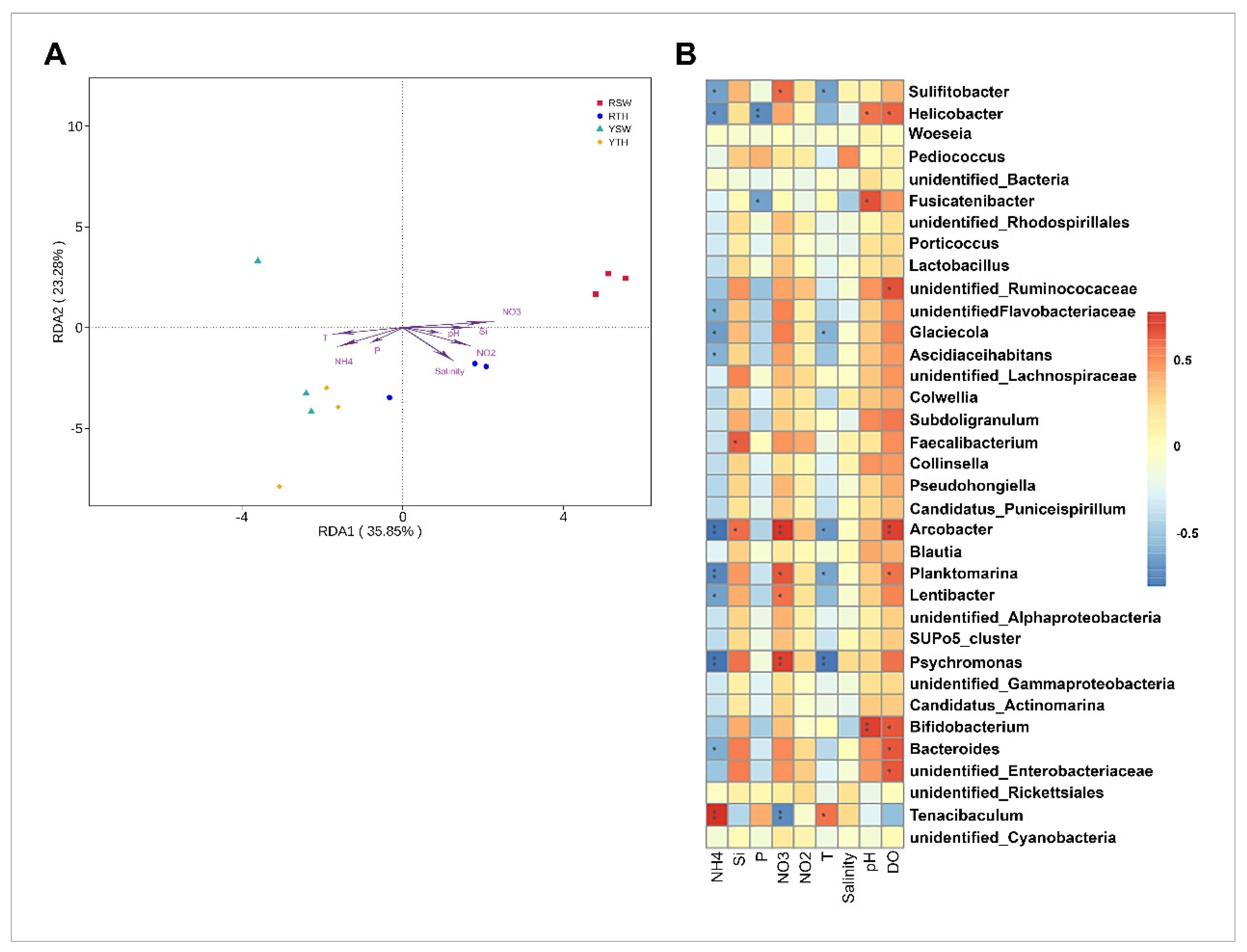
3.5. Linkage between Bacterial Community Structures and Seawater Properties
4. Discussion
4.1. Bacterial Community Structure and Abundance
4.2. Effects of Abiotic Factors on Community Structure
4.3. Functional Profiles Of Bacterial Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tseng, C.K. Marine Phycoculture in China. In International Seaweed Symposium, 10th ed.; Levrig, T., Ed.; De Gruyter: Berlin, Germany; Boston, MA, USA, 1981; pp. 123–152. [Google Scholar] [CrossRef]
- Wells, M.L.; Potin, P.; Craigie, J.S.; Raven, J.A.; Merchant, S.S.; Helliwell, K.E.; Smith, A.G.; Camire, M.E.; Brawley, S.H. Algae as Nutritional and Functional Food Sources: Revisiting Our Understanding. J. Appl. Phycol. 2017, 29, 949–982. [Google Scholar] [CrossRef]
- Stoyneva-Gärtner, M.P.; Uzunov, B.A. An Ethnobiological Glance on Globalization Impact on the Traditional Use of Algae and Fungi as Food in Bulgaria. J. Nutr. Food Sci. 2015, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Pereira, L. A Review of the Nutrient Composition of Selected Edible Seaweeds. In Ecology, Nutrient Composition and Medicinal Uses, 1st ed.; Pomin, V.H., Ed.; Nova Science Pub.: Coimbra, Portugal, 2011; pp. 15–47. [Google Scholar]
- FAO. Fishery and Aquaculture Statistics. Global Aquaculture Production. In FAO Fisheries and Aquaculture Department; FAO: Rome, Italy, 2019; Available online: http://www.fao.org/fishery/ (accessed on 18 September 2019).
- Levine, I.A.; Sahoo, D. Porphyra: Harvesting Gold from the Sea; I.K. International Pub. House: New Delhi, India, 2010. [Google Scholar]
- Campbell, A.H.; Marzinelli, E.M.; Gelber, J.; Steinberg, P.D. Spatial Variability of Microbial Assemblages Associated with a Dominant Habitat-Forming Seaweed. Front. Microbiol. 2015, 6, 230. [Google Scholar] [CrossRef] [PubMed]
- Egan, S.; Thomas, T. Editorial for: Microbial symbiosis of marine sessile hosts- diversity and function. Front. Microbiol. 2015, 6, 585. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.; Vanderklift, M.A.; Poore, A.G.B. Strong Effects of Herbivorous Amphipods on Epiphyte Biomass in a Temperate Seagrass Meadow. Mar. Ecol. Prog. Ser. 2011, 442, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Bulleri, F.; Chapman, M.G. The Introduction of Coastal Infrastructure as a Driver of Change in Marine Environments. J. Appl. Ecol. 2010, 47, 26–35. [Google Scholar] [CrossRef]
- Goecke, F.; Labes, A.; Wiese, J.; Imhoff, J.F. Chemical Interactions between Marine Macroalgae and Bacteria. Mar. Ecol. Prog. Ser. 2010, 409, 267–299. [Google Scholar] [CrossRef]
- Provasoli, L.; Pintner, I.J. Bacteria Induced Polymorphism in an Axenic Laboratory Strain of Ulva Lactuca (Chlorophyceae) 1. J. Phycol. 1980, 16, 196–201. [Google Scholar] [CrossRef]
- Pedersen, M. The Demand for Iodine and Bromine of Three Marine Brown Algae Grown in Bacteria-Free Cultures. Physiol. Plant. 1969, 22, 680–685. [Google Scholar] [CrossRef]
- Kingman, A.R.; Moore, J. Isolation, Purification and Quantitation of Several Growth Regulating Substances in Ascophyllum Nodosum (Phaeophyta). BOT MAR 1982, 25, 149–154. [Google Scholar] [CrossRef]
- Saga, N. A New Method for Pure Culture of Macroscopic Algae, the One Step Selection Method. Jpn. J. Phycol. 1982, 30, 40–45. [Google Scholar]
- Matsuo, Y.; Imagawa, H.; Nishizawa, M.; Shizuri, Y. Isolation of an Algal Morphogenesis Inducer from a Marine Bacterium. Science 2005, 307, 1598. [Google Scholar] [CrossRef]
- Ghaderiardakani, F.; Coates, J.C.; Wichard, T. Bacteria-Induced Morphogenesis of Ulva Intestinalis and Ulva Mutabilis (Chlorophyta): A Contribution to the Lottery Theory. FEMS Microbiol. Ecol. 2017, 93, fix094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, A.; Costa, R.; Wichard, T. Morphogenesis of Ulva Mutabilis (Chlorophyta) Induced by Maribacter Species (Bacteroidetes, Flavobacteriaceae). Bot Mar. 2017, 60, 197–206. [Google Scholar] [CrossRef]
- Fries, L. Polysiphonia Urceolata in Axenic Culture. Nature 1964, 202, 110. [Google Scholar] [CrossRef]
- Aydlett, M. Examining the Microbiome of Porphyra Umbilicalis in the North Atlantic. Honors Thesis, University of Maine, Orono, Maine, 2019. Available online: https://digitalcommons.library.umaine.edu/honors/570 (accessed on 4 September 2019).
- Yan, Y.-W.; Yang, H.-C.; Tang, L.; Li, J.; Mao, Y.-X.; Mo, Z.-L. Compositional Shifts of Bacterial Communities Associated with Pyropia Yezoensis and Surrounding Seawater Co-Occurring with Red Rot Disease. Front. Microbiol. 2019, 10, 1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhi, Y.; He, Y.; Ren, Z.; Chen, H.; Yang, R. Changes in Phycospheric and Environmental Microbes Associated with an Outbreak of Yellow Spot Disease on Pyropia Yezoensis. Aquaculture 2020, 529, 735651. [Google Scholar] [CrossRef]
- Wang, J.; Mao, Y.; Du, G.; Li, X.; Tang, X. On Microbial Community of Pyropia Haitanensis by Metagenomic Analysis. J. Oceanol. Limnol. 2020, 1–12. [Google Scholar] [CrossRef]
- Zhang, R.; Chang, L.; Xiao, L.; Zhang, X.; Han, Q.; Li, N.; Egan, S.; Wang, G. Diversity of the Epiphytic Bacterial Communities Associated with Commercially Cultivated Healthy and Diseased Saccharina Japonica during the Harvest Season. J. Appl. Phycol. 2020, 32, 2071–2080. [Google Scholar] [CrossRef]
- Gamolo, L.; Besagas, R.L. Microbiological Profile of Cultured Seaweeds Kappaphycus Alvarezii. Int. J. Biosci. 2018, 13, 140–145. [Google Scholar] [CrossRef]
- Khan, S.; Mao, Y.; Zou, D.; Riaz, S.; Qiu, L.; Li, N. Molecular Analysis of Microbiota Composition and Alterations in Pyropia Yezoensis Infected with Red Rot Disease. Indian J. Geo-Mar. Sci. 2018, 47, 558–566. [Google Scholar]
- Osman, M.-A.; Neoh, H.-M.; Ab Mutalib, N.-S.; Chin, S.-F.; Jamal, R. 16S RRNA Gene Sequencing for Deciphering the Colorectal Cancer Gut Microbiome: Current Protocols and Workflows. Front. Microbiol. 2018, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Fang, W.Y.; Shan, Y.Y.; Chen, H.M.; Sun, X.; Ye, Y. Genetic Diversity of Epiphytic Bacteria in Porphyra Yezoensis. Acta Oceanol. Sin. 2008, 30, 161–168. [Google Scholar]
- Shen, M.; Yang, R.; Luo, Q.; Wang, S.; Ren, J. Microbial diversity of Pyropia haitanensis phycosphere during cultivation. Wei Sheng Wu Xue Bao 2013, 53, 1087–1102. [Google Scholar]
- Eaton, A.D.; Clesceri, L.S.; Rice, E.W.; Greenberg, A.E.; Franson, M.H.A. STANDARD Methods for the Examination of Water and Wastewater; American Public Health Association: Washington, DC, USA, 2005. [Google Scholar]
- Magoč, T.; Salzberg, S.L. FLASH: Fast Length Adjustment of Short Reads to Improve Genome Assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-Filtering Vastly Improves Diversity Estimates from Illumina Amplicon Sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S RRNA Sequence Formation and Detection in Sanger and 454-Pyrosequenced PCR Amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.; Knight, R.; et al. Predictive Functional Profiling of Microbial Communities Using 16S RRNA Marker Gene Sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Algina, J.; Keselman, H.J. Comparing Squared Multiple Correlation Coefficients: Examination of a Confidence Interval and a Test Significance. Psychol. Methods 1999, 4, 76–83. [Google Scholar] [CrossRef]
- Hartman, W.H.; Ye, R.; Horwath, W.R.; Tringe, S.G. A Genomic Perspective on Stoichiometric Regulation of Soil Carbon Cycling. ISME J. 2017, 11, 2652–2665. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, H.; de Sousa, T.; Santos, J.P.; Sousa, A.G.G.; Teixeira, C.; Monteiro, M.R.; Salgado, P.; Mucha, A.P.; Almeida, C.M.R.; Torgo, L.; et al. Potential of Dissimilatory Nitrate Reduction Pathways in Polycyclic Aromatic Hydrocarbon Degradation. Chemosphere 2018, 199, 54–67. [Google Scholar] [CrossRef]
- LeBrun, E.S.; Kang, S. A Comparison of Computationally Predicted Functional Metagenomes and Microarray Analysis for Microbial P Cycle Genes in a Unique Basalt-Soil Forest. F1000Research 2018, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-J.; Shao, Y.; Li, Y.-J.; Lin, L.-A.; Chen, Y.; Tian, W.; Li, B.-L.; Li, Y.-Y. Rhizosphere Bacterial Community Structure and Predicted Functional Analysis in the Water-Level Fluctuation Zone of the Danjiangkou Reservoir in China during the Dry Period. Int. J. Environ. Res. Public Health 2020, 17, 1266. [Google Scholar] [CrossRef] [Green Version]
- Bulleri, F.; Benedetti-Cecchi, L.; Acunto, S.; Cinelli, F.; Hawkins, S.J. The Influence of Canopy Algae on Vertical Patterns of Distribution of Low-Shore Assemblages on Rocky Coasts in the Northwest Mediterranean. J. Exp. Mar. Bio. Ecol. 2002, 267, 89–106. [Google Scholar] [CrossRef]
- Schiel, D.R.; Lilley, S.A. Gradients of Disturbance to an Algal Canopy and the Modification of an Intertidal Community. Mar. Ecol. Prog. Ser. 2007, 339, 1–11. [Google Scholar] [CrossRef]
- Alongi, D.M. Coastal Ecosystem Processes, 1st ed.; CRC Press: Boca Raton, FL, USA, 2019; pp. 1–10. [Google Scholar]
- Lachnit, T.; Meske, D.; Wahl, M.; Harder, T.; Schmitz, R. Epibacterial Community Patterns on Marine Macroalgae Are Host-Specific but Temporally Variable: Epiphytic Communities on Macroalgae. Environ. Microbiol. 2011, 13, 655–665. [Google Scholar] [CrossRef]
- Albakosh, M.A.; Naidoo, R.K.; Kirby, B.; Bauer, R. Identification of Epiphytic Bacterial Communities Associated with the Brown Alga Splachnidium Rugosum. J. Appl. Phycol. 2016, 28, 1891–1901. [Google Scholar] [CrossRef]
- Selvarajan, R.; Sibanda, T.; Venkatachalam, S.; Ogola, H.J.O.; Christopher Obieze, C.; Msagati, T.A. Distribution, Interaction and Functional Profiles of Epiphytic Bacterial Communities from the Rocky Intertidal Seaweeds, South Africa. Sci. Rep. 2019, 9, 19835. [Google Scholar] [CrossRef]
- Nakanishi, K.; Nishijima, M.; Nishimura, M.; Kuwano, K.; Saga, N. Bacteria that induce morphogenesis in Ulva Pertusa (Chlorophyta) grown under axenic conditions. J. Phycol. 1996, 32, 479–482. [Google Scholar] [CrossRef]
- Lee, Y.K.; Lee, J.H.; Lee, H.K. Microbial Symbiosis in Marine Sponges. J. Microbiol. 2001, 39, 254–264. [Google Scholar]
- Lesser, M.P.; Mazel, C.H.; Gorbunov, M.Y.; Falkowski, P.G. Discovery of Symbiotic Nitrogen-Fixing Cyanobacteria in Corals. Science 2004, 305, 997–1000. [Google Scholar] [CrossRef]
- Marshall, K.; Joint, I.; Callow, M.E.; Callow, J.A. Effect of Marine Bacterial Isolates on the Growth and Morphology of Axenic Plantlets of the Green Alga Ulva Linza. Microb. Ecol. 2006, 52, 302–310. [Google Scholar] [CrossRef]
- Rosenberg, E.; Koren, O.; Reshef, L.; Efrony, R.; Zilber-Rosenberg, I. The Role of Microorganisms in Coral Health, Disease and Evolution. Nat. Rev. Microbiol. 2007, 5, 355–362. [Google Scholar] [CrossRef]
- Yakimov, M.M.; Cappello, S.; Crisafi, E.; Tursi, A.; Savini, A.; Corselli, C.; Scarfi, S.; Giuliano, L. Phylogenetic Survey of Metabolically Active Microbial Communities Associated with the Deep-Sea Coral Lophelia Pertusa from the Apulian Plateau, Central Mediterranean Sea. Deep Sea Res. Part 1 Oceanogr. Res. Pap. 2006, 53, 62–75. [Google Scholar] [CrossRef]
- Longford, S.R.; Tujula, N.A.; Crocetti, G.R.; Holmes, A.J.; Holmström, C.; Kjelleberg, S.; Steinberg, P.D.; Taylor, M.W. Comparisons of Diversity of Bacterial Communities Associated with Three Sessile Marine Eukaryotes. Aquat. Microb. Ecol. 2007, 48, 217–229. [Google Scholar] [CrossRef]
- Reis, A.M.M.; Araújo, S.D., Jr.; Moura, R.L.; Francini-Filho, R.B.; Pappas, G., Jr.; Coelho, A.M.A.; Krüger, R.H.; Thompson, F.L. Bacterial Diversity Associated with the Brazilian Endemic Reef Coral Mussismilia Braziliensis. J. Appl. Microbiol. 2009, 106, 1378–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.W.; Schupp, P.J.; Dahllöf, I.; Kjelleberg, S.; Steinberg, P.D. Host Specificity in Marine Sponge-Associated Bacteria, and Potential Implications for Marine Microbial Diversity: Host Specificity and Diversity in Marine Bacteria. Environ. Microbiol. 2004, 6, 121–130. [Google Scholar] [CrossRef]
- Egan, S.; Thomas, T.; Kjelleberg, S. Unlocking the Diversity and Biotechnological Potential of Marine Surface Associated Microbial Communities. Curr. Opin. Microbiol. 2008, 11, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.; Thomas, T.; Lewis, M.; Steinberg, P.; Kjelleberg, S. Composition, Uniqueness and Variability of the Epiphytic Bacterial Community of the Green Alga Ulva Australis. ISME J. 2011, 5, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonthond, G.; Bayer, T.; Krueger-Hadfield, S.A.; Barboza, F.R.; Nakaoka, M.; Valero, M.; Wang, G.; Künzel, S.; Weinberger, F. How Do Microbiota Associated with an Invasive Seaweed Vary across Scales? Mol. Ecol. 2020, 29, 2094–2108. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Reddy, C.R.K. Seaweed-Microbial Interactions: Key Functions of Seaweed-Associated Bacteria. FEMS Microbiol. Ecol. 2014, 88, 213–230. [Google Scholar] [CrossRef]
- Oliveira, L.S.; Gregoracci, G.B.; Silva, G.G.Z.; Salgado, L.T.; Amado Filho, G.; Alves-Ferreira, M.; Thompson, F.L. Transcriptomic Analysis of the Red Seaweed Laurencia Dendroidea (Florideophyceae, Rhodophyta) and Its Microbiome. BMC Genom. 2012, 13, 487. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.W. Bacterial Communities on Macroalgae. In Ecological Studies; Springer: Berlin/Heidelberg, Germany, 2012; pp. 189–201. [Google Scholar] [CrossRef]
- Hollants, J.; Decleyre, H.; Leliaert, F.; De Clerck, O.; Willems, A. Life without a Cell Membrane: Challenging the Specificity of Bacterial Endophytes within Bryopsis (Bryopsidales, Chlorophyta). BMC Microbiol. 2011, 11, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, L.; Pusceddu, A.; Stabili, L.; Alifano, P.; Fraschetti, S. Potential Effects of an Invasive Seaweed (Caulerpa Cylindracea, Sonder) on Sedimentary Organic Matter and Microbial Metabolic Activities. Sci. Rep. 2017, 7, 12113. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, P.; Huang, Y.; Wang, J.; Garrido, I.; Leiva, S. Phylogeny and Bioactivity of Epiphytic Gram-Positive Bacteria Isolated from Three Co-Occurring Antarctic Macroalgae. Antonie Van Leeuwenhoek 2018, 111, 1543–1555. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusch, D.B.; Halpern, A.L.; Sutton, G.; Heidelberg, K.B.; Williamson, S.; Yooseph, S.; Wu, D.; Eisen, J.A.; Hoffman, J.M.; Remington, K.; et al. The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol. 2007, 5, e77. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Stingl, U. Molecular Diversity and Ecology of Microbial Plankton. Nature 2005, 437, 343–348. [Google Scholar] [CrossRef]
- Nowlan, J.P.; Lumsden, J.S.; Russell, S. Advancements in Characterizing Tenacibaculum Infections in Canada. Pathogens 2020, 9, 1029. [Google Scholar] [CrossRef]
- Teramoto, M.; Zhai, Z.; Komatsu, A.; Shibayama, K.; Suzuki, M. Genome Sequence of the Psychrophilic Bacterium Tenacibaculum Ovolyticum Strain Da5A-8 Isolated from Deep Seawater. Genome Announc. 2016, 4, e00644-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.-X.; Yu, P.; Mu, D.-S.; Liu, Y.; Du, Z.-J. Tenacibaculum Agarivorans Sp. Nov., an Agar-Degrading Bacterium Isolated from Marine Alga Porphyra Yezoensis Ueda. Int. J. Syst. Evol. Microbiol. 2017, 67, 5139–5143. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Colon, C.; Joyner, J. Sediment Microbiomes Associated with Critical Habitat of the Juvenile American Horseshoe Crab; Limulus Polyphemus. Glob. J. Environ. Sci. Manag. 2020, 6, 309–322. [Google Scholar] [CrossRef]
- Suzuki, M.; Nakagawa, Y.; Harayama, S.; Yamamoto, S. Phylogenetic Analysis and Taxonomic Study of Marine Cytophaga-like Bacteria: Proposal for Tenacibaculum Gen. Nov. with Tenacibaculum Maritimum Comb. Nov. and Tenacibaculum Ovolyticum Comb. Nov., and Description of Tenacibaculum Mesophilum Sp. Nov. and Tenacibaculum Amylolyticum Sp. Nov. Int. J. Syst. Evol. Microbiol. 2001, 51 Pt 5, 1639–1652. [Google Scholar] [CrossRef]
- Habib, C.; Houel, A.; Lunazzi, A.; Bernardet, J.-F.; Olsen, A.B.; Nilsen, H.; Toranzo, A.E.; Castro, N.; Nicolas, P.; Duchaud, E. Multilocus Sequence Analysis of the Marine Bacterial Genus Tenacibaculum Suggests Parallel Evolution of Fish Pathogenicity and Endemic Colonization of Aquaculture Systems. Appl. Environ. Microbiol. 2014, 80, 5503–5514. [Google Scholar] [CrossRef] [Green Version]
- Quéméneur, M.; Bel Hassen, M.; Armougom, F.; Khammeri, Y.; Lajnef, R.; Bellaaj-Zouari, A. Prokaryotic Diversity and Distribution along Physical and Nutrient Gradients in the Tunisian Coastal Waters (South Mediterranean Sea). Front. Microbiol. 2020, 11, 593540. [Google Scholar] [CrossRef] [PubMed]
- Anandan, R.; Dharumadurai, D.; Manogaran, G.P. An Introduction to Actinobacteria. In Actinobacteria-Basics and Biotechnological Applications; InTech: London, UK, 2018; pp. 3–37. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.; Chang, B.X.; Morris, R.M. Cultivation of a Chemoautotroph from the SUP05 Clade of Marine Bacteria That Produces Nitrite and Consumes Ammonium. ISME J. 2017, 11, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Nimnoi, P.; Pongsilp, N. Marine Bacterial Communities in the Upper Gulf of Thailand Assessed by Illumina Next-Generation Sequencing Platform. BMC Microbiol. 2020, 20, 19. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Jia, Q.; Chen, X.; Köllner, T.G.; Bhattacharya, D.; Wong, G.K.-S.; Gershenzon, J.; Chen, F. Terpene Biosynthesis in Red Algae Is Catalyzed by Microbial Type but Not Typical Plant Terpene Synthases. Plant Physiol. 2019, 179, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Egan, S.; Harder, T.; Burke, C.; Steinberg, P.; Kjelleberg, S.; Thomas, T. The Seaweed Holobiont: Understanding Seaweed-Bacteria Interactions. FEMS Microbiol. Rev. 2013, 37, 462–476. [Google Scholar] [CrossRef] [Green Version]
- Vandendriessche, S.; Vincx, M.; Degraer, S. Floating Seaweed and the Influences of Temperature, Grazing and Clump Size on Raft Longevity—a Microcosm Study. J. Exp. Mar. Biol. Ecol. 2007, 343, 64–73. [Google Scholar] [CrossRef]
- Macaya, E.C.; Boltana, S.; Hinojosa, I.A.; Macchiavello, J.E.; Valdivia, N.A.; Vasquez, N.R.; Buschmann, A.H.; Vasquez, J.A.; Alonso, J.M.; Thiel, M. Presence of Sporophylls in floating kelp rafts of Macrocystis spp. (Phaeophyceae) along the Chillean pacific coast. J. Phycol. 2005, 41, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Vandendriessche, S.; De Keersmaecker, G.; Vincx, M.; Degraer, S. Food and Habitat Choice in Floating Seaweed Clumps: The Obligate Opportunistic Nature of the Associated Macrofauna. Mar. Biol. 2006, 149, 1499–1507. [Google Scholar] [CrossRef] [Green Version]
- Juneja, A.; Ceballos, R.; Murthy, G. Effects of Environmental Factors and Nutrient Availability on the Biochemical Composition of Algae for Biofuels Production: A Review. Energies 2013, 6, 4607–4638. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Gawad, K.M.; Hifney, A.F.; Issa, A.A.; Gomaa, M. Spatio-Temporal, Environmental Factors, and Host Identity Shape Culturable-Epibiotic Fungi of Seaweeds in the Red Sea. Egypt. Hydrobiol. 2014, 740, 37–49. [Google Scholar] [CrossRef]
- Jokiel, P.L. Solar Ultraviolet Radiation and Coral Reef Epifauna. Science 1980, 207, 1069–1071. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.S.; Gąbka, M. The Influence of Abiotic Factors on the Bloom-Forming Alga Ulva Flexuosa (Ulvaceae, Chlorophyta): Possibilities for the Control of the Green Tides in Freshwater Ecosystems. J. Appl. Phycol. 2018, 30, 1405–1416. [Google Scholar] [CrossRef] [Green Version]
- Lemay, M.A.; Martone, P.T.; Keeling, P.J.; Burt, J.M.; Krumhansl, K.A.; Sanders, R.D.; Wegener Parfrey, L. Sympatric Kelp Species Share a Large Portion of Their Surface Bacterial Communities. Environ. Microbiol. 2018, 20, 658–670. [Google Scholar] [CrossRef]
- Vonshak, A.; Torzillo, G. Environmental Stress Physiology. In Handbook of Microalgae Culture. Biotechnology and Applied Phycology; Richmond, A., Ed.; Blackwell Science Ltd.: Hoboken, NJ, USA, 2004; pp. 57–82. [Google Scholar] [CrossRef]
- Joint, I.; Smale, D.A. Marine Heatwaves and Optimal Temperatures for Microbial Assemblage Activity. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [Green Version]
- Dang, H.; Lovell, C.R. Microbial Surface Colonization and Biofilm Development in Marine Environments. Microbiol. Mol. Biol. Rev. 2016, 80, 91–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gestel, N.C.; Ducklow, H.W.; Bååth, E. Comparing Temperature Sensitivity of Bacterial Growth in Antarctic Marine Water and Soil. Glob. Chang. Biol. 2020, 26, 2280–2291. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, H.; Gatesoupe, F.-J.; She, R.; Lin, Q.; Yan, X.; Li, J.; Li, X. Bacterial Signatures of “Red-Operculum” Disease in the Gut of Crucian Carp (Carassius Auratus). J. Phycol. 2017, 74, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Shilova, I.N.; Mills, M.M.; Robidart, J.C.; Turk-Kubo, K.A.; Björkman, K.M.; Kolber, Z.; Rapp, I.; van Dijken, G.L.; Church, M.J.; Arrigo, K.R.; et al. Differential Effects of Nitrate, Ammonium, and Urea as N Sources for Microbial Communities in the North Pacific Ocean: N Effects on Microbial Communities. Limnol. Oceanogr. 2017, 62, 2550–2574. [Google Scholar] [CrossRef]
- Cirri, E.; Pohnert, G. Algae− Bacteria Interactions That Balance the Planktonic Microbiome. New Phytol. 2019, 223, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Černá, M. Seaweed Proteins and Amino Acids as Nutraceuticals. Adv. Food Nutr. Res. 2011, 64, 297–312. [Google Scholar] [CrossRef]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-Ya, K.; Omura, S.; Cane, D.E.; Ikeda, H. Terpene Synthases Are Widely Distributed in Bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Schulz, S.; Dickschat, J.S. Bacterial Volatiles: The Smell of Small Organisms. Nat. Prod. Rep. 2007, 24, 814–842. [Google Scholar] [CrossRef]
- Bassler, B.L. Small Talk: Cell-to-Cell Communication in Bacteria. Cell 2002, 109, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Waters, C.M.; Bassler, B.L. Quorum Sensing: Cell-to-Cell Communication in Bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [Green Version]
- Mangwani, N.; Dash, H.R.; Chauhan, A.; Das, S. Bacterial Quorum Sensing: Functional Features and Potential Applications in Biotechnology. J. Mol. Microbiol. Biotechnol. 2012, 22, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lyu, Y.; Richlen, M.; Anderson, D.M.; Cai, Z. Quorum Sensing Is a Language of Chemical Signals and Plays an Ecological Role in Algal-Bacterial Interactions. CRC Crit. Rev. Plant. Sci. 2016, 35, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Decho, A.W.; Frey, R.L.; Ferry, J.L. Chemical Challenges to Bacterial AHL Signaling in the Environment. Chem. Rev. 2011, 111, 86–99. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Dong, Y.-H. Quorum Sensing and Signal Interference: Diverse Implications: Signal Interference. Mol. Microbiol. 2004, 53, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.W.; Radax, R.; Steger, D.; Wagner, M. Sponge-Associated Microorganisms: Evolution, Ecology, and Biotechnological Potential. Microbiol. Mol. Biol. Rev. 2007, 71, 295–347. [Google Scholar] [CrossRef] [Green Version]
- Håkansson, S.; Galyov, E.E.; Rosqvist, R.; Wolf-Watz, H. The Yersinia YpkA Ser./Thr Kinase Is Translocated and Subsequently Targeted to the Inner Surface of the HeLa Cell Plasma Membrane. Mol. Microbiol. 1996, 20, 593–603. [Google Scholar] [CrossRef]
- Galyov, E.E.; Håkansson, S.; Forsberg, A.; Wolf-Watz, H. A Secreted Protein Kinase of Yersinia Pseudotuberculosis Is an Indispensable Virulence Determinant. Nature 1993, 361, 730–732. [Google Scholar] [CrossRef]
- Archambaud, C.; Gouin, E.; Pizarro-Cerda, J.; Cossart, P.; Dussurget, O. Translation Elongation Factor EF-Tu is a Target for Stp, a Serine-Threonine Phosphatase Involved in Virulence of Listeria Monocytogenes: EF-Tu, a Target for the Listeria Phosphatase Stp. Mol. Microbiol. 2005, 56, 383–396. [Google Scholar] [CrossRef]
- Lima, A.; Durán, R.; Schujman, G.E.; Marchissio, M.J.; Portela, M.M.; Obal, G.; Cerveñansky, C. Serine/Threonine Protein Kinase PrkA of the Human Pathogen Listeria Monocytogenes: Biochemical Characterization and Identification of Interacting Partners through Proteomic Approaches. J. Proteom. 2011, 74, 1720–1734. [Google Scholar] [CrossRef] [PubMed]
- Alber, T. Signaling Mechanisms of the Mycobacterium Tuberculosis Receptor Ser./Thr Protein Kinases. Curr. Opin. Struct. Biol. 2009, 19, 650–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molle, V.; Kremer, L. Division and Cell Envelope Regulation by Ser/Thr Phosphorylation: Mycobacterium Shows the Way. Mol. Microbiol. 2010, 75, 1064–1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, C.; Yang, H.; Mushegian, A.; Jin, S. A Novel Serine/Threonine Protein Kinase Homologue of Pseudomonas Aeruginosa Is Specifically Inducible within the Host Infection Site and Is Required for Full Virulence in Neutropenic Mice. J. Bacteriol. 1998, 180, 6764–6768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristich, C.J.; Wells, C.L.; Dunny, G.M. A Eukaryotic-Type Ser/Thr Kinase in Enterococcus Faecalis Mediates Antimicrobial Resistance and Intestinal Persistence. Proc. Natl. Acad. Sci. USA 2007, 104, 3508–3513. [Google Scholar] [CrossRef] [Green Version]
- Beltramini, A.M.; Mukhopadhyay, C.D.; Pancholi, V. Modulation of Cell Wall Structure and Antimicrobial Susceptibility by a Staphylococcus Aureus Eukaryote-like Serine/Threonine Kinase and Phosphatase. Infect. Immun. 2009, 77, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Truong-Bolduc, Q.C.; Ding, Y.; Hooper, D.C. Posttranslational Modification Influences the Effects of MgrA on NorA Expression in Staphylococcus Aureus. J. Bacteriol. 2008, 190, 7375–7381. [Google Scholar] [CrossRef] [Green Version]
- Truong-Bolduc, Q.C.; Hooper, D.C. Phosphorylation of MgrA and Its Effect on Expression of the NorA and NorB Efflux Pumps of Staphylococcus Aureus. J. Bacteriol. 2010, 192, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- Canova, M.J.; Molle, V. Bacterial Serine/Threonine Protein Kinases in Host-Pathogen Interactions. J. Biol. Chem. 2014, 289, 9473–9479. [Google Scholar] [CrossRef] [Green Version]
- Croft, M.T.; Lawrence, A.D.; Raux-Deery, E.; Warren, M.J.; Smith, A.G. Algae Acquire Vitamin B 12 through a Symbiotic Relationship with Bacteria. Nature 2005, 438, 90–93. [Google Scholar] [CrossRef]
- Kazamia, E.; Czesnick, H.; Nguyen, T.T.V.; Croft, M.T.; Sherwood, E.; Sasso, S.; Smith, A.G. Mutualistic Interactions between Vitamin B12-dependent Algae and Heterotrophic Bacteria Exhibit Regulation. Environ. Microbiol. 2012, 14, 1466–1476. [Google Scholar] [CrossRef]
- Danchin, A.; Braham, S. Coenzyme B12 Synthesis as a Baseline to Study Metabolite Contribution of Animal Microbiota. Microb. Biotechnol. 2017, 10, 688–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goecke, F.; Thiel, V.; Wiese, J.; Labes, A.; Imhoff, J.F. Algae as an Important Environment for Bacteria–Phylogenetic Relationships among New Bacterial Species Isolated from Algae. Phycologia 2013, 52, 14–24. [Google Scholar] [CrossRef]
- Florez, J.Z.; Camus, C.; Hengst, M.B.; Buschmann, A.H. A Functional Perspective Analysis of Macroalgae and Epiphytic Bacterial Community Interaction. Front. Microbiol. 2017, 8, 2561. [Google Scholar] [CrossRef] [PubMed]
- Grueneberg, J.; Engelen, A.H.; Costa, R.; Wichard, T. Macroalgal Morphogenesis Induced by Waterborne Compounds and Bacteria in Coastal Seawater. PLoS ONE 2016, 11, e0146307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, D.; Webb, J.S.; Holmström, C.; Case, R.; Low, A.; Steinberg, P.; Kjelleberg, S. Low Densities of Epiphytic Bacteria from the Marine Alga Ulva Australis Inhibit Settlement of Fouling Organisms. Appl. Environ. Microbiol. 2007, 73, 7844–7852. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Xiao, T.; Pang, S.; Liu, M.; Yue, H. Isolation and Identification of Bacteria Associated with the Surfaces of Several Algal Species. Chin. J. Oceanol. Limnol. 2009, 27, 487–492. [Google Scholar] [CrossRef]
- Zozaya-Valdes, E.; Egan, S.; Thomas, T. A Comprehensive Analysis of the Microbial Communities of Healthy and Diseased Marine Macroalgae and the Detection of Known and Potential Bacterial Pathogens. Front. Microbiol. 2015, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Barbeyron, T.; Martin, R.; Portetelle, D.; Michel, G.; Vandenbol, M. The Cultivable Surface Microbiota of the Brown Alga Ascophyllum Nodosum Is Enriched in Macroalgal-Polysaccharide-Degrading Bacteria. Front. Microbiol. 2015, 6, 1487. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environmental Variables | YSW | RSW | p-Value |
|---|---|---|---|
| DO (ppm) | 15.86 ± 0.161 | 16.39 ± 0.44 | 0.123 |
| Temperature (°C) | 6.8 ± 0.205 | 5.9 ± 0.200 | 0.005 * |
| Salinity (%) | 3.28 ± 0.097 | 3.33 ± 0.118 | 0.684 |
| pH | 6.90 ± 0.200 | 7.09 ± 0.262 | 0.639 |
| NH4 (μmol/L) | 2.527 ± 0.260 | 0.85 ± 0.429 | 0.004 * |
| Si (μmol/L) | 5.39 ± 0.263 | 6.30 ± 0.921 | 0.175 |
| NO3 (μmol/L) | 4.70 ± 0.806 | 11.301 ± 0.901 | 0.001 * |
| NO2 (μmol/L) | 0.751 ± 0.032 | 0.836 ± 0.069 | 0.130 |
| Phosphate (μmol/L) | 0.177 ± 0.015 | 0.178 ± 0.009 | 0.911 |
| Sample Groups | Whole-Tree PD | Chao1, ACE | Simpson | Shannon | Observed Species |
|---|---|---|---|---|---|
| RTH-YTH | 0.091 | 0.071 | 0.244 | 0.071 | 0.047 * |
| RSW-YSW | 0.155 | 0.071 | 0.047 * | 0.071 | 0.024 * |
| RTH-RSW | 0.053 | 0.029 * | 0.002 * | 0.004 * | 0.012 * |
| YTH-YSW | 0.031 * | 0.029 * | 0.492 | 0.854 | 0.024 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, A.; Khurshid, A.; Tang, X.; Wang, J.; Khan, T.U.; Mao, Y. Structural and Functional Impacts of Microbiota on Pyropia yezoensis and Surrounding Seawater in Cultivation Farms along Coastal Areas of the Yellow Sea. Microorganisms 2021, 9, 1291. https://doi.org/10.3390/microorganisms9061291
Ahmed A, Khurshid A, Tang X, Wang J, Khan TU, Mao Y. Structural and Functional Impacts of Microbiota on Pyropia yezoensis and Surrounding Seawater in Cultivation Farms along Coastal Areas of the Yellow Sea. Microorganisms. 2021; 9(6):1291. https://doi.org/10.3390/microorganisms9061291
Chicago/Turabian StyleAhmed, Arsalan, Anam Khurshid, Xianghai Tang, Junhao Wang, Tehsin Ullah Khan, and Yunxiang Mao. 2021. "Structural and Functional Impacts of Microbiota on Pyropia yezoensis and Surrounding Seawater in Cultivation Farms along Coastal Areas of the Yellow Sea" Microorganisms 9, no. 6: 1291. https://doi.org/10.3390/microorganisms9061291