1. Introduction
With future limitations in the availability of fossil resources looming and increasing environmental concerns, the use of lignocellulosic biomass for the production of bio-based fuels and chemicals has increasingly attracted global attention. However, due to the recalcitrant structure of lignocellulose, its enzymatic deconstruction is still a major scientific and practical challenge and remains a topic demanding further research efforts [
1]. Lignocellulose is mainly comprised of three primary polymers: cellulose (40.6–51.2%), hemicelluloses (28.5–37.2%) and a lower fraction of lignin (13.6–28.1%) [
2], which form an intricate matrix.
Various plant cell wall polymers contain arabinose, which is the second most abundant pentose in nature [
3]. Arabinose-containing polysaccharides such as arabinoxylan (AX), arabinan and arabinogalactan (AG) are found as major components of hemicelluloses and pectic substances, respectively. AX has a linear backbone of β-1,4-linked
d-xylopyranosyl units, which are randomly single or double substituted at the O-3 and/or O-2 position by
l-arabinose moieties [
4]. Some of the arabinose residues are linked to ferulic acid by ester bonds and the formation of ferulate dimers creates arabinoxylan-arabinoxylan cross-links [
5]. Arabinans and arabinogalactans are constituents of pectic substances [
6]. Arabinan forms side-chains in the rhamnogalacturonan I or occurs as a free polysaccharide. It consists of a linear α-1,5-linked
l-arabinofuranosyl polymer as the backbone, which can be decorated with arabinofuranosyl residue at O-3 and/or O-2 position, such as in sugar beet arabinan (SBA) [
7], without its side chains this polysaccharide is called debranched arabinan (DA). AG consists of β-1,6-glycosidically linked
d-galactopyranose residues as the backbone, which is modified by side chains of α-arabinose, β-galactose and 4-
O-methylglucuronic acid [
8]. All these arabinose-containing polymers of hemicellulose and pectic substances play an important role in the crosslinking within the plant cell wall structure and thus increase the complexity of plant cell wall architecture.
For complete deconstruction of arabinose-containing polysaccharides, microorganisms produce various enzymes with different substrate and cleavage specificities, such as endo-β-1,4-xylanase, β-1,4-xylosidase, endoarabinanase, α-
l-arabinofuranosidase or β-1,3-galactosidase [
9,
10]. Glycoside hydrolase family 43 (GH43) encompasses various structurally related enzymes with an inverting hydrolysis mechanism involved in the degradation of arabinose-containing carbohydrates [
11]. For example, endoarabinanase is capable of hydrolyzing the α-1,5-linked
l-arabinofuranosyl backbone of arabinans to produce arabino-oligosaccharides, exo-α-1,5-arabinanases are active on the non-reducing end of arabinan chain releasing arabinobiose or arabinotriose [
12,
13], whereas α-
l-arabinofuranosidase cleaves off side chain decorations in arabino-oligosaccharides or arabinose-containing polysaccharides, e.g., with xylan, galactan or arabinan backbones, thus liberating
l-arabinose [
14]. However, arabinofuranosyl di-substitutions such as found on xylose residues of certain xylans withstand cleavage by most known arabinofuranosidases [
15]. Besides GH43, arabinofuranosidases are also found as members of GH3, GH51, GH54 and GH62 [
11]. According to their cleavage specificities in arabinoxylan degradation, arabinofuranosidases are grouped into different types [
16]: As the most common type of arabinofuranosidases, AXHs-m remove arabinose moieties from 1,2- or 1,3-monosubstituted main-chain xylose residues, which can be found in most of the mentioned GH families [
10,
17], while AXHs-d cleave off either α-1,2- or α-1,3-linked arabinofuranosyl side chains from double-substituted main-chain xylose residues, additional numbers can be used to describe a more specific cleavage mode, such as AXH-d3 refers to the enzymes that only cleave off 1,3-linked arabinofuranosyl residue from double substituted Xylp motifs [
18,
19,
20] and AXHs-m,d finally remove arabinofuranosyl substitutions from monosubstituted and double-substituted xylose residues [
18,
21]. Some α-
l-arabinofuranosidases are also able to cleave off terminal α-
l-arabinofuranosyl residues from decorated or linear arabinan [
21].
Strain N2K1 is a novel cellulolytic organism, recently described as
Hungateiclostridium mesophilum sp. nov. and shortly reclassified as
Acetivibrio mesophilus, which was isolated from a mesophilically operated biogas plant fed with maize silage [
22,
23]. The physiological characterization of this organism revealed its high potential for degrading recalcitrant substrates from plant biomass (wheat arabinoxylan, oat spelt xylan, sugar beet pulp and cellulose), so this organism can be regarded as an attractive source of enzymes for biotechnological applications. The 4.04 Mbp genome assembly of strain N2K1
T was annotated to contain a wide variety of glycoside hydrolase genes for the depolymerization of the major polysaccharides in plant cell walls. However, while gene annotation mostly considers sequence similarity or secondary structures, the true enzymatic function of the putative proteins often cannot be predicted accurately, even if high sequence similarity to other enzymes is evident [
24].
In the genome sequence of A. mesophilus N2K1, we found an approximately 26 kbp large gene cluster of colinearly arranged genes for glycoside hydrolases, transport proteins and regulatory proteins putatively involved in hemicellulose and pectin substrate degradation. Here we mainly focus on the enzymes with the potential for degradation of arabinose-containing heteroxylan or pectic arabinan encoded in this gene cluster. Specifically, we chose two arabinofuranosidases belonging to the GH families GH43 and GH51 and designated AmAraf43 and AmAraf51, respectively, for characterization and comparison. Furthermore, we also explored the synergistic action modes of these enzymes on SBA and AX degradation by the combination with endoactive polysaccharide hydrolases.
2. Materials and Methods
2.1. Bacterial Strains and Plasmids
Acetivibrio mesophilus (basonym.
Hungateiclostridium mesophilum) strain N2K1
T is a novel anaerobic, mesophilic and cellulolytic bacterium isolated from a biogas fermenter, as recently described by Rettenmaier et al. [
22,
23]. Genomic DNA from this strain (NCBI Reference Sequence ID: 85920) served as a template for gene amplification. The
E. coli strain XL1-Blue (Stratagene, La Jolla, CA, USA) was used for general cloning purposes, propagation of recombinant plasmids and cloning. The
E. coli strain ArcticExpress (DE3) was used as a host for protein production by gene expression from pET24c vectors. The bacteria were cultivated in lysogeny broth (LB) or grown on LB agar plates supplemented with the appropriate antibiotics. Kanamycin was used at 50 μg mL
−1 and gentamycin at 20 μg mL
−1.
2.2. Gene Cloning and Protein Purification
Genomic DNA of strain N2K1 served as a template for amplification of the genes with GenBank (NCBI) numbers RXE58498.1 (AmAraf51) and RXE58512.1 (AmAraf43), which encode a putative α-1,5-l-arabinofuranosidase (AmAraf51) and a putative endo-1,4-β-xylanase (AmAraf43), respectively. The primers used in PCR reactions include AmAraf51_f, ATGCATCATCACCATCACCATAAAAAAGCCAGAATGACC, and AmAraf51_r, CAGTGGTGGTGGTGGTGGTGCGTTATTTCCCCAGTCGAATTAC, for the gene RXE58498.1. The generated PCR product then served as a template for a second PCR reaction with the same reverse primer but a different forward primer, ACTTTAAGAAGGAGATATACAATGCATCATCACCATCACCAT, to introduce six in-frame histidine codons at the 5′ end of the target ORF. The primers for amplification of the target gene RXE58512.1 encoding AmAraf43 were AmAraf43_f, CTTTAAGAAGGAGATATACATATGTTATTTACAAAAAAAGCC, and AmAraf43_r, TCAGTGGTGGTGGTGGTGGTGCGCTTCAATAAAAGTAAACCAATTC, which included the sequence for the in-frame 3′-extension of the target ORF with six histidine codons.
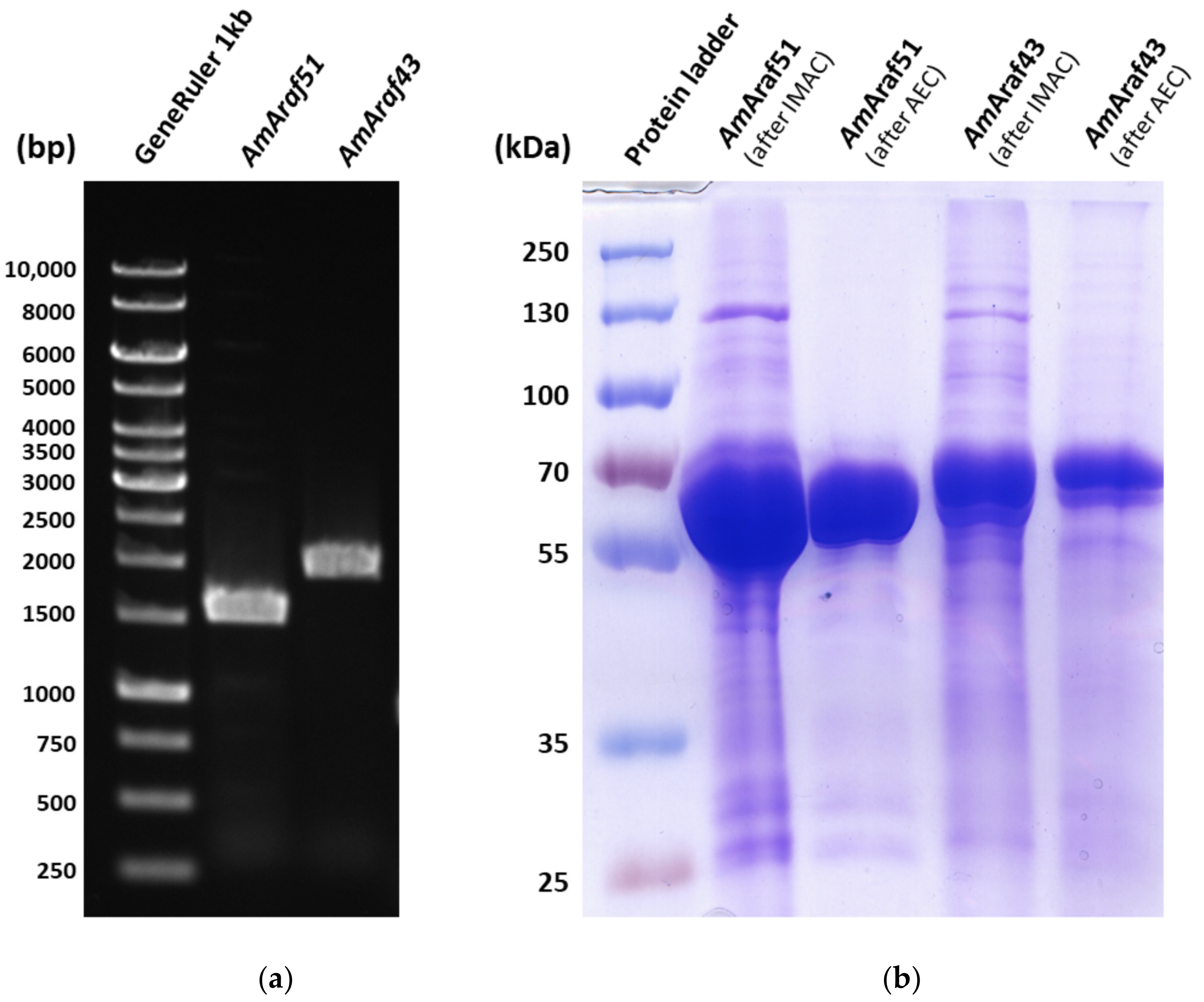
The amplicons were then cloned in NdeI/XhoI linearized pET24c vector by the Gibson Assembly Master Mix (New England Biolabs). All the recombinant plasmids were subsequently transformed into competent cells of E. coli strain XL1 by heat shock transformation. Correct plasmid construction was verified by restriction analysis and DNA sequencing before their introduction into expression host E. coli ArcticExpress. An ArcticExpress transformant carrying the recombinant plasmid was inoculated into 5 mL of lysogenic broth (LB) medium supplemented with 50 μg mL−1 kanamycin and 20 μg mL−1 gentamycin and grown aerobically at 37 °C overnight. The overnight culture was inoculated into 1 L of LB and incubated with shaking (180 rpm) at 30 °C for 6 h before addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and incubation at 13 °C, 250 rpm for 24 h. The cells were harvested and disrupted by sonication with a Hielscher UP200S apparatus (Hielscher Ultrasonics GmbH, Teltow, Germany) for 5 min (50% amplitude, 0.4 s cycle), and the cell-free crude extract was prepared by centrifuging the cell lysate at 13,400× g, at 4 °C for 30 min followed by filtration through a 0.45 µm filter. AmAraf51 and AmAraf43 were purified by immobilized metal affinity chromatography (IMAC) using a nickel column (Machery-Nagel Protino® Ni-TED 2000, Fischer Scientific GmbH, Schwerte, Germany). The elution fraction containing the soluble protein was further purified by using an ÄKTA pure 25L1 FPLC system (GE Healthcare Life Sciences, Amersham, UK) equipped with a HiTrapTM 1 mL QFF column, using a linear gradient from 0 to 1 M NaCl. The size and purity of proteins were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by pooling the fractions containing pure enzyme. The protein concentration was determined with the Bradford assay with bovine serum albumin (BSA) as a standard.
2.3. Enzyme Assays
Arabinoxylan (from wheat flour, insoluble; WAX-I), arabinoxylan (from wheat flour for the reducing sugar assay; WAX-RS), arabinan (from sugar beet pulp; SBA), debranched arabinan (from sugar beet pulp, arabinose:galactose:rhamnose = 71:26:3; DA), linear arabinan (from sugar beet pulp, arabinose: galactose:rhamnose:galacturonic acid = 85.2:7.6:1.5:5.7; LA), arabino-oligosaccharides (AOS), xylo-oligosaccharides (XOS) and arabino-xylo-oligosaccharides (AXOS) were purchased from Megazyme (Wicklow, Ireland), Beechwood xylan were purchased from SERVA Electrophoresis GmbH (Heidelberg, Germany). WAX-I had an Araf:Xylp ratio of 36:51 while still maintaining ferulic acid crosslinks during the process of substrate extraction, while WAX-RS had an Araf:Xylp ratio of 38:62 with diferulate bridges mostly broken due to alkaline treatment during substrate extraction [
21,
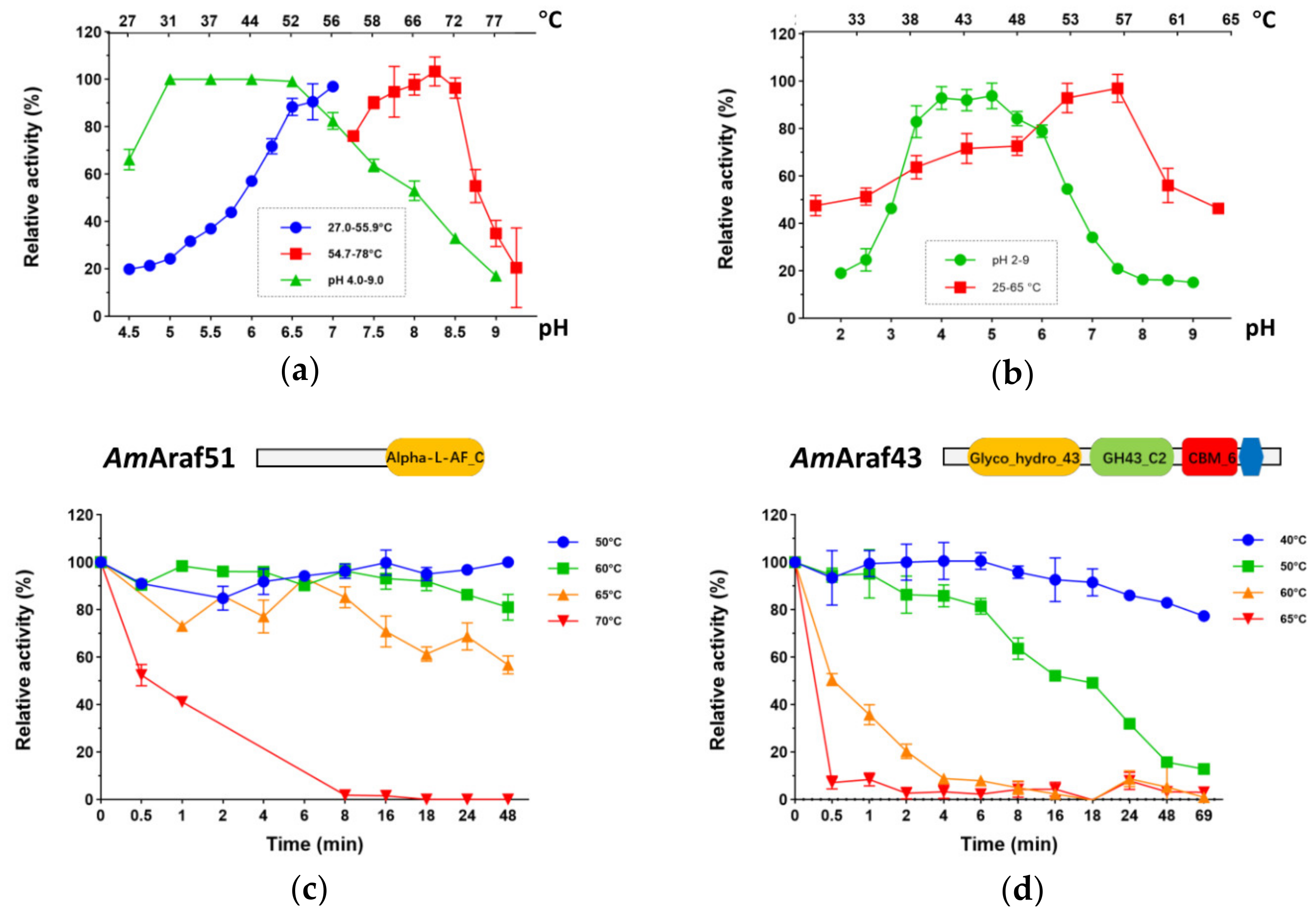
25]. All insoluble substrates used in this study were washed with Milli-Q water followed by centrifugation before starting the enzyme assay. The optimal temperature (at pH 6.0 for both
AmAraf51and
AmAraf43) and pH (at 70 °C for
AmAraf51 or 57 °C for
AmAraf43) of the enzymes were tested in the pH range between pH 2.0 and 9.0 (pH adjusted at 60 °C) and the temperature range between 25 and 90 °C, respectively, with appropriately diluted enzyme in 25 mM citrate-phosphate buffer (citric acid, Na
2HPO
4, 50 mM NaCl) using
p-nitrophenyl-α-
l-arabinofuranoside (
pNP-AF) (Megazyme) as the substrate at a final concentration of 0.2 mM. The reaction was stopped after 20 min by adding two volumes of 1 M Na
2CO
3, followed by a photometrical measurement at 405 nm. The resistance of the enzymes against thermoinactivation was measured by comparing their changes in activity against
pNP-AF, before and after heat treatment for various time spans (0 h, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h,16 h, 18 h, 24 h and 48 h).
The specific activities of the enzymes on arabinose/xylose-based polysaccharide substrates were measured using the substrates at a final concentration of 5 g L
−1 in 25 mM citrate-phosphate buffer at their optimal pH and appropriate enzyme concentrations (between 360 nM to 2 µM). The reactions were performed at optimal temperature with 600 rpm shaking on an Eppendorf thermomixer. At specific intervals, aliquots of 200 μL were withdrawn and cooled on ice. For the insoluble substrate, aliquots were centrifuged at 4 °C for 5 min at 13,000 rpm, then the supernatant was transferred to new tubes for further analysis. The reducing ends liberated during the enzymatic hydrolysis reactions were quantified with the 3,5-dinitrosalicylic acid (DNS) reducing sugar method [
26]. To this end, 50 µL samples of the enzyme reactions were mixed with 75 µL of DNS reagent, followed by incubation for 10 min at 95 °C in 96 well PCR plates in a thermocycler. The products were then transferred into wells of 96 well microtiter plates and the absorbance were measured at 540 nm. All assays were performed in triplicate. One unit of activity was defined as the amount of enzyme needed to release 1 µmol of
l-arabinose equivalent per minute.
Kinetic parameters were determined on SBA and washed DA for AmAraf51 and on WAX-RS for AmAraf43 by incubating at 60 °C and at 50 °C, respectively, under shaking at 600 rpm using an Eppendorf thermomixer, 200 µL of reaction mixtures containing 25 mM citrate-phosphate buffer, various concentrations of substrates (45 g L−1, 40 g L−1, 35 g L−1, 30 g L−1, 25 g L−1, 15 g L−1, 10 g L−1, 7.5 g L−1, 5 g L−1, 2.5 g L−1 and 1 g L−1 for SBA and DA; 25 g L−1, 20 g L−1, 15 g L−1, 10 g L−1, 7.5 g L−1, 5 g L−1, 2.5 g L−1 and 1 g L−1 for WAX-RS) and different concentrations of the enzyme (final concentration of 30 µg mL−1, 20 µg mL−1 and 10 µg mL−1 for AmAraf51 and 70 µg mL−1, 50 µg mL−1 and 30 µg mL−1 for AmAraf43), after two hours incubation at 60 °C (AmAraf51) or 50 °C (AmAraf43), aliquots were cooled on ice and centrifuged at 4 °C for 5 min at 13,000 rpm. Then, 50 µL of supernatant was mixed with 75 µL of DNS reagent in 96 well plates for the DNS assay and absorbance readout at 540 nm as described above. All assays were performed in triplicate.
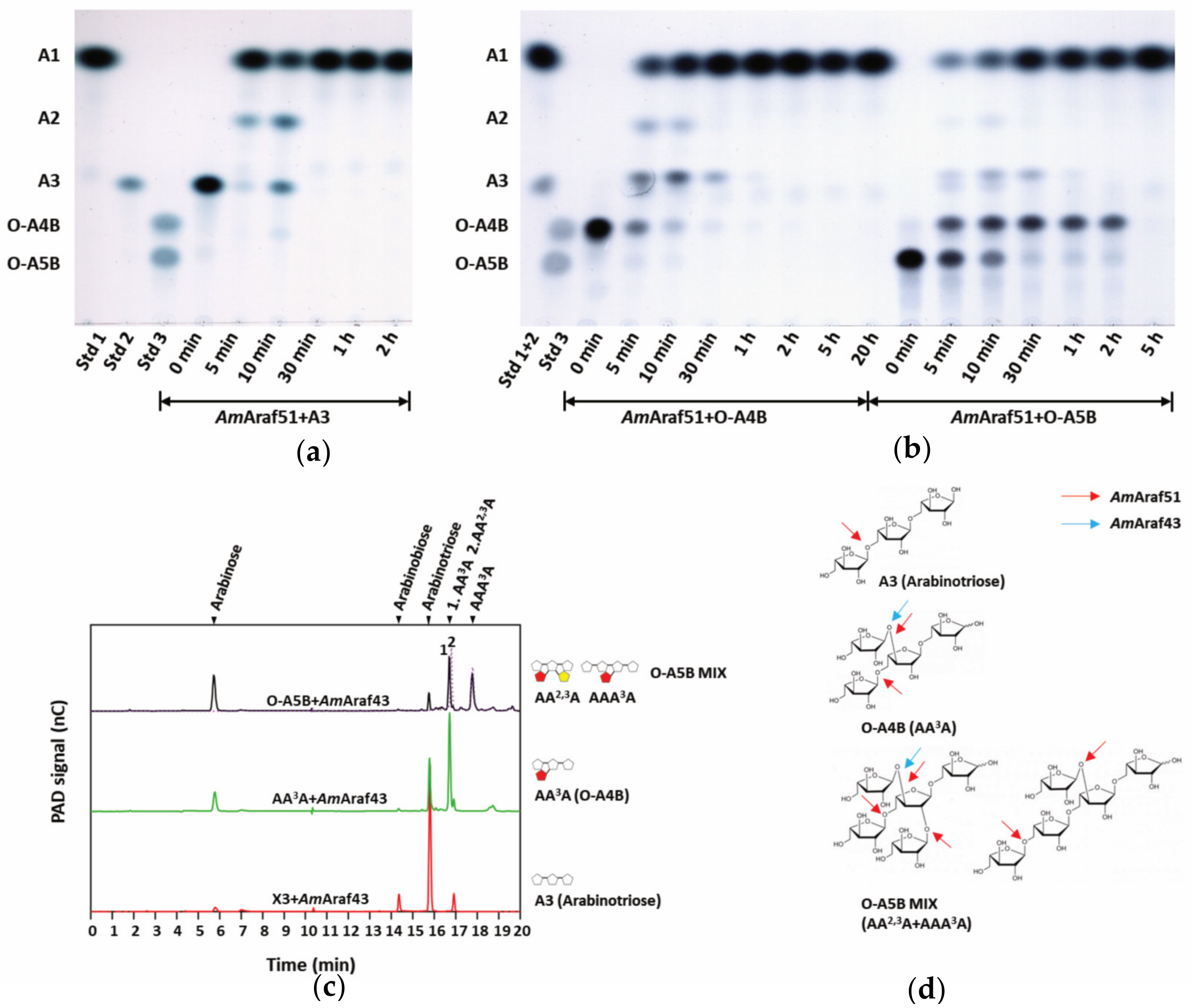
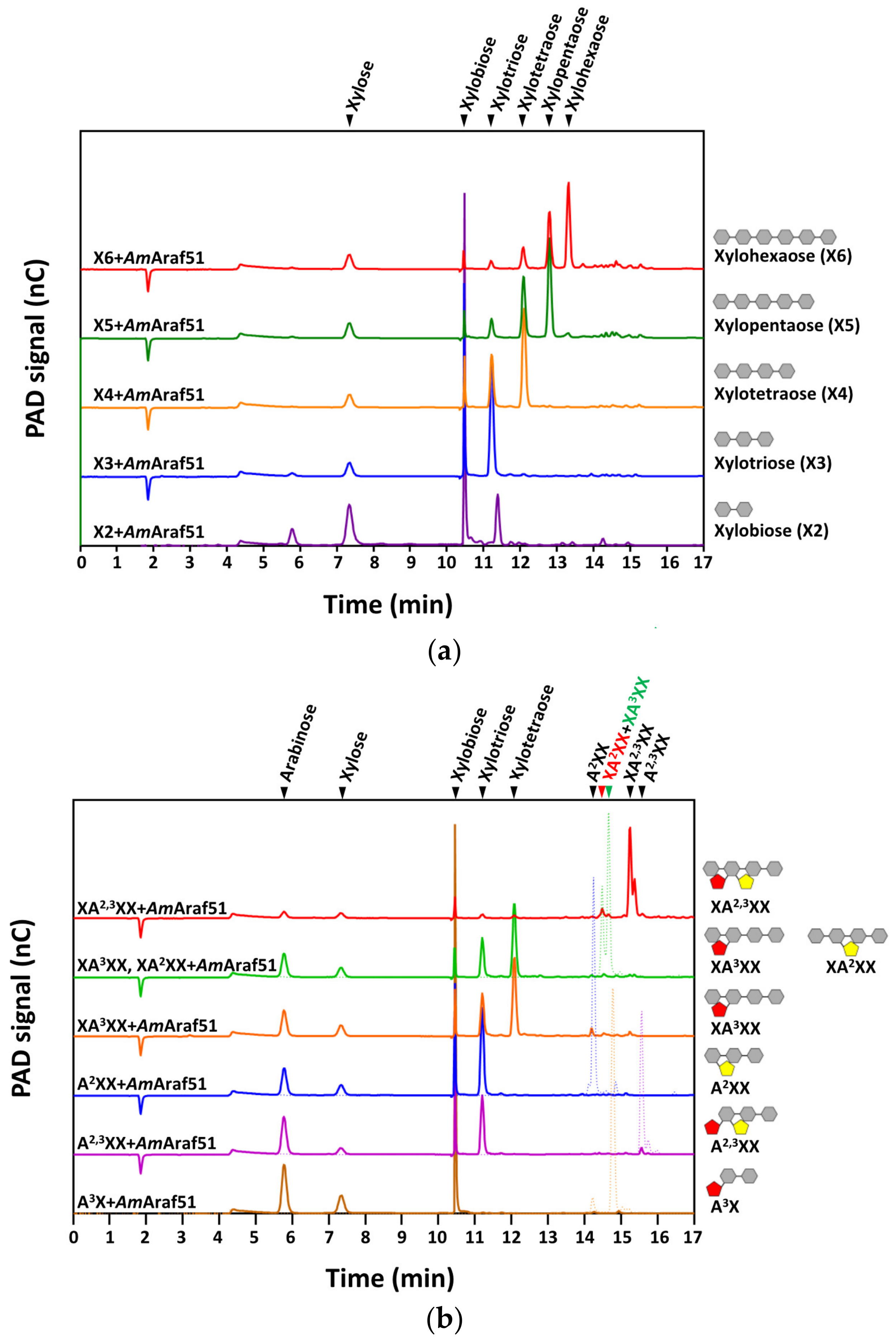
To explore the enzymatic action mode and preference towards the specific side chains of arabino-/xylo-oligosaccharides, the oligosaccharides, including A3, O-A4B, O-A5B, O-XTR, O-XTE, O-XPE, O-XHE, O-XBI, O-A3X, O-AX3, O-XAXX MIX, O-XA3XX, O-A2X3 and O-XA23XX (the structures of the oligosaccharides are shown in
Table S1), were individually incubated in a thermocycler at a final substrate concentration of 0.5 g L
−1 with 85 nM
AmAraf51 or 82 nM
AmAraf43 at pH 6.0, 60 °C or pH 5.0, 40 °C, respectively, for 24 h. For the reactions with arabino-oligosaccharide substrates (A3, O-A4B and O-A5B), 10 µL of the reaction mixtures were collected at specific intervals (5 min, 10 min, 30 min, 1 h, 5 h and 20 h), then the reactions were terminated by incubation at 100 °C for 10 min. The hydrolysis products were identified by high performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD) using a Dionex ICS 3000 SP system equipped with a CarboPac PA1 column (4 mm × 250 mm) and a PA1 precolumn (4 mm × 50 mm) as described by Angelov et al. [
27]. Analytes were injected and the analysis was performed at 30 °C at a flow rate of 1 mL/min. Separation was achieved in 100 mM sodium hydroxide using an increasing sodium acetate gradient within 45 min. The program was set as follows: 0–10 min: a linear gradient from 0 to 100 mM sodium acetate, 10–30 min: a linear gradient from 100 to 800 mM sodium acetate and 30–45 min: 0 mM sodium acetate. Thin layer chromatography (TLC) was also performed for the qualitative analysis of the hydrolysis products released from different oligosaccharides. For TLC analysis, 3 µL of reaction mixtures were loaded on silica gel 60 TLC plates (Merck, Germany) and separated with three consecutive runs in saturated TLC chambers using a solvent mixture of chloroform:acetate:water (6:7:1,
v/
v/
v), fully drying the plates between each run. The analytes separated on the plate were visualized by spraying with 2.5 mL staining solution containing 1% aniline (
v/
v) and 1% (
w/
v) diphenylamine in acetone mixed with 0.1 volume 85% H
3PO
4 in a DESAGA ChromaJet DS20 TLC spray chamber (Sarstedt, Nümbrecht, Germany), followed by heating for 10 min at 120 °C.
For examining the influence of metal ions (CuCl2, NiSO4, FeCl2 4H2O, ZnCl2, CoCl2, MnCl2 4H2O, CaCl2 2H2O, NaCl, KCl, MgCl2 6H2O and MgSO4), denaturants (SDS and urea), chelator (EDTA) and arabinose, these substances (except arabinose) were incubated with enzyme (0.56 µM AmAraf51 or 0.16 µM AmAraf43) at final concentrations of 10 mM, 5 mM and 1 mM in 25 mM citrate phosphate buffer at room temperature for 2 h, including negative controls, which contained only individual additives or only an enzyme. The effect of arabinose on both enzymes was tested by incubating 67 mM, 133 mM, 266 mM or 533 mM arabinose with 0.56 µM AmAraf51 or 0.16 µM AmAraf43 for 2 h, respectively, and a negative control using water instead of arabinose was included. Upon incubation, 2 mM pNP-AF was added to the reaction mixtures and residual activities were measured with the colorimetric pNP assay described above.
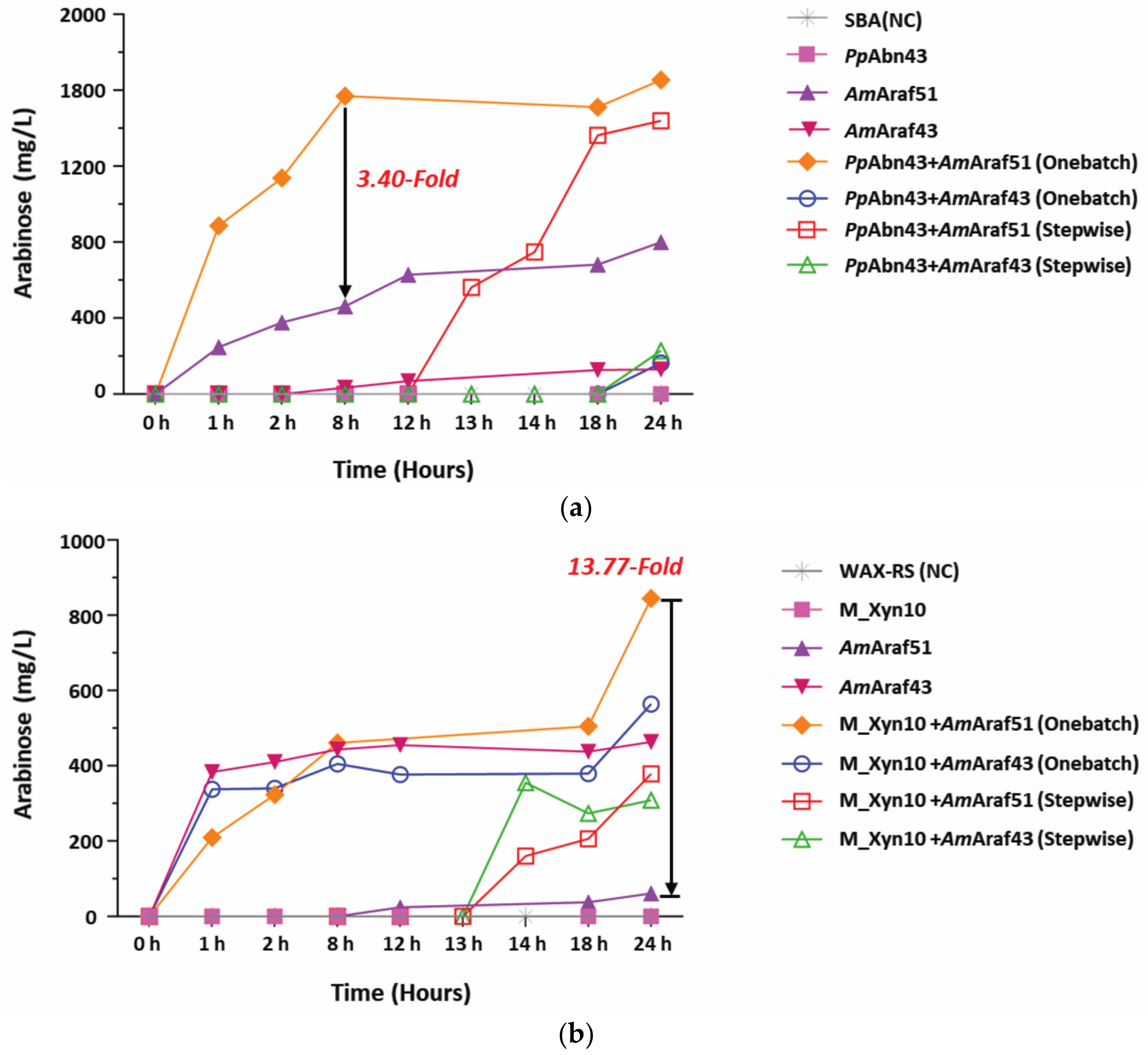
In order to determine synergistic effects and hydrolysis yields obtained during SBA degradation and WAX-RS degradation, the two α-l-arabinofuranosidases AmAraf51 and AmAraf43 were combined with two endoactive enzymes, PpAbn43 (arabinanase from Paenibacillus polymyxa DSM 292) and M_Xyn10 (xylanase from metagenomic library screening), two enzymes available in our laboratory. At a substrate concentration of 5 g L−1 of SBA or WAX-RS, different combinations of the endo- and exoacting enzymes were incubated in 25 mM citrate phosphate buffer (pH 5.5) at 50 °C for 24 h. For stepwise enzyme digestions, the endocleaving enzyme (18 µg each, corresponding to 0.9 µM PpAbn43 and 2.9 µM M_Xyn10) was first incubated with a substrate (SBA or WAX-RS, respectively) for 12 h, followed by heat inactivation by boiling for 10 min and thereafter adding the exo-acting enzyme (0.3 µM AmAraf51 or AmAraf43) and incubation for an additional 12 h. For simultaneous enzyme digestions, reaction mixtures with SBA were incubated for 24 h with either 0.3 µM AmAraf51 plus 0.9 µM PpAbn43 or 0.3 µM AmAraf43 plus 0.9 µM PpAbn43 and reaction mixtures containing WAX-RS were incubated for 24 h with either 0.3 µM AmAraf51 plus 2.9 µM M_Xyn10 or 0.3 µM AmAraf43 plus 2.9 µM M_Xyn10. Arabinose and xylose liberated at different time points (0 h, 1 h, 2 h, 6 h, 12 h, 13 h, 14 h, 18 h and 24 h) were quantified by HPAEC-PAD, referring to a standard curve obtained from a series of different arabinose/xylose concentrations (200 mg L−1, 100 mg L−1, 50 mg L−1, 25 mg L−1 and 12.5 mg L−1).
4. Discussion
In nature, bacteria degrade recalcitrant polymeric carbohydrates by carbohydrate-active enyzmes [
11], which include glycoside hydrolases, polysaccharide lyases, carbohydrate esterases and glycosyl transferases plus a range of auxiliary enzymes [
30]. Often, CAZyme-encoding genes are organized in clusters, frequently containing genes for sugar transport and regulation, in particular in bacteria specialized on the degradation of complex plant cell wall polysaccharides [
31]. Some saccharolytic bacteria from the group of clostridia have evolved special multifunctional protein complexes named cellulosomes, which integrate numerous cellulases, hemicellulases and accessory enzymes assembled on non-catalytic scaffold proteins and form highly efficient (hemi)cellulolytic systems [
32]. For example, the genome of
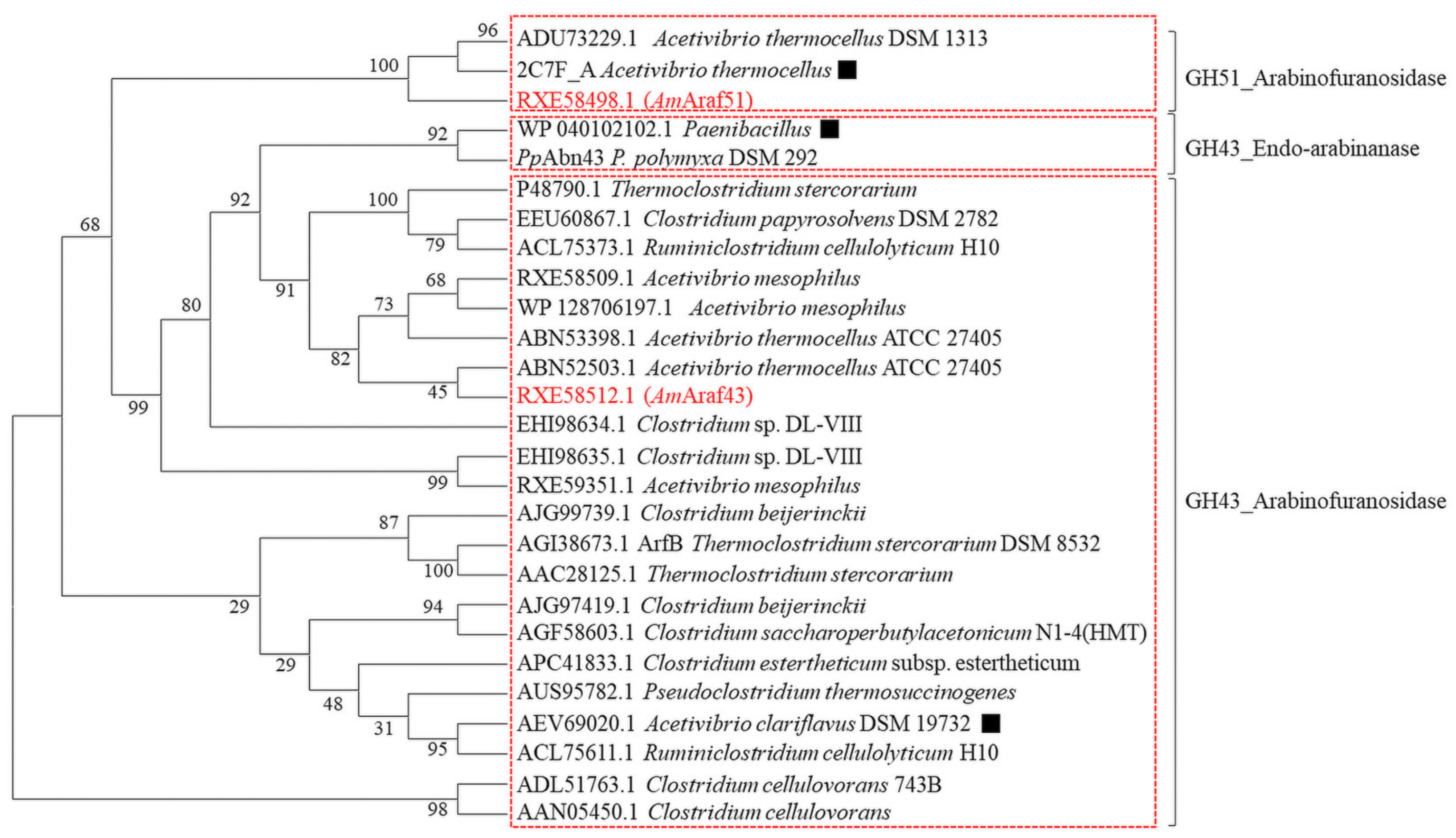
Ruminiclostridium cellulolyticum contains a 32-kb
xyl-doc gene cluster encoding 14 putative cellulosome enzymes, which are predicted to be involved in hemicellulose degradation [
21], four of them were characterized to have α-
l-arabinofuranosidase activity. In the genome of the recently isolated
Acetivibrio mesophilus (basonym
Hungateiclostridium mesophilum) strain N2K1 [
22], the gene cluster we focused on here, contains two genes annotated to encode for α-
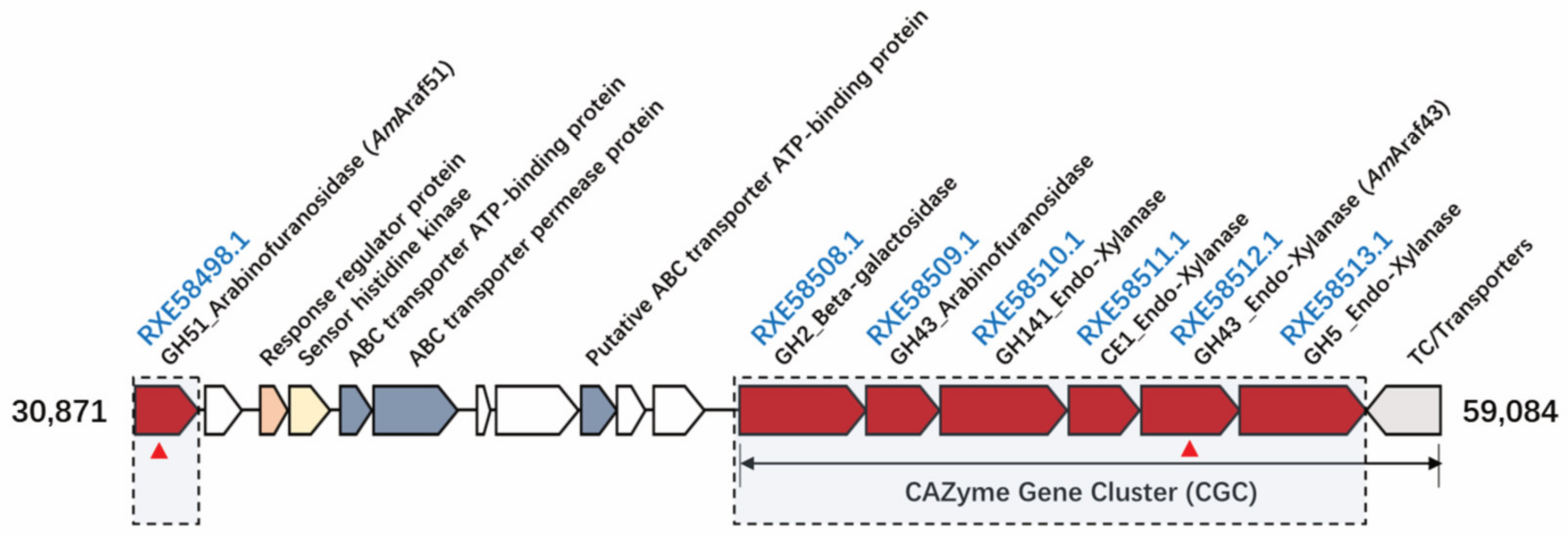
l-arabinofuranosidases (RXE58498.1 and RXE58509.1, but the latter without corresponding enzymatic activity) and other genes predicted to be involved in hemicellulose degradation including genes for a putative β-galactosidase (RXE58508.1) and four putative endoxylanases arrayed downstream of gene RXE58509.1 (including RXE58510.1, RXE58511.1, RXE58512.1 and RXE58513.1), all in the same orientation (
Figure 1). Upstream of these genes, a gene for a putative GH51 α-
l-arabinofuranosidase (
AmAraf51) without a signal peptide was found (RXE58498.1) separated from the other CAZyme genes by several genes encoding transport and regulatory proteins.
Due to the predicted functions, it can be suggested that the CAZymes gene cluster is involved in hemicellulose breakdown and uptake of the breakdown products, which is in agreement with the ability of strain N2K1 to decompose complex lignocellulosic substrates such as cellulose, wheat arabinoxylan, oat spelt xylan and sugar beet pulp [
22]. All the genes between RXE58508.1 and RXE58513.1 contain C-terminal dockerin domain- and
N-terminal signal peptide-encoding sequences (signal peptides predicted with SignalP-5.0), indicating that these genes encode extracellular cellulosomal proteins. Since strain N2K1 is a poor utilizer of externally added monosaccharides [
22], the co-occurrence of transport protein-encoding genes in the hemicellulose degradation gene cluster suggests that oligosaccharides are transported into the cell before being further hydrolyzed to monomeric sugars. Based on the lack of a signal peptide,
AmAraf51 (product of RXE58498.1) can be assumed to be an intracellular protein and may be involved in the hydrolysis of arabinose-containing oligosaccharides upon their internalization into the cytoplasm, like other cytosolic arabinofuranosidases [
33]. However, a (partially) extracellular role could still be possible, since for example the extracellular proteome analysis of a
B. subtilis strain revealed a significant number of proteins without a signal peptide in the culture supernatant [
34], which may have been released by alternative secretion mechanisms or by bacterial cell lysis [
35].
Arabinofuranosidases play an important role in the saccharification of arabinose-containing substrates, including arabinan and arabinoxylan. WAX-RS and WAX-I have almost the same Ara
f/Xyl
p ratio of 38:62 and 36:51, respectively, but WAX-I degradation is more difficult due to the presence of ferulic acid cross-links and the acetylation of xylose residues, whereas WAX-RS is extracted under alkaline conditions resulting in the removal of the ferulic acid residues [
21,
25]. Sugar beet arabinan as used in our study contains arabinofuranose residues attached to around 60% of O-3 positions of the α-1,5-linked arabinan backbone units and less frequently with α-1,2 arabinofuranose [
36], while debranched arabinan exhibits a backbone with a very low degree of substitution.
AmAraf51 and
AmAraf43 have totally opposite preferences toward arabinan or arabinoxylan substrates:
AmAraf51 has a much higher specific activity against arabinan substrates, especially debanched arabinan (decreasing order of activities on linear arabinan (LA) > debranched arabinan (DA) > sugar beet arabinan (SBA)), demonstrating that
AmAraf51 has a strong ability to cleave α-1,5-arabinofuranosyl linkages of the arabinan backbone. On the other hand, its activity, measured against the arabinoxylans WAX-I and WAX-RS, was at the detection limit of the assay used. The substrate (arabinan) backbone preference of
AmAraf51 also seems to be reflected by its hydrolysis reactions with oligosaccharides (also see below).
AmAraf51 efficiently cleaved off the di-arabinosyl substitutions on interior residues of an arabinose-based backbone (AA
2,3A), but not of a xylopyranosyl backbone (XA
2,3XX) (
Figure 5b and
Figure 6b). Since
AmAraf51 furthermore cleaves off the arabinosyl branches of AOS, this enzyme is proposed to play a role in the utilization of arabinan and AOS by strain N2K1.
AmAraf43 exhibited a converse order of substrate preferences compared with
AmAraf51, with the highest specific activity towards WAX-RS, which was followed by WAX-I, while almost no activity was detected with pectic arabinan.
AmAraf43 lacks the ability to cleave the arabinan backbone. This enzyme, which was originally annotated to be a putative endoxylanase, also did not cleave the xylan backbone, yielding only arabinose but no xylo-oligosaccharides as hydrolysis products after incubation with WAX. Thus,
AmAraf43 can be classified as an exo-α-
l-arabinofuranosidase, not surprisingly demonstrating that automated annotation can be misleading with regard to GH43 enzymes. Analogously, the enzyme encoded by gene
axb8 from
A. thermocellus (
C. thermocellum) strain B8 (GH43_29) was predicted to be an exoarabinofuranosidase but only released xylose instead of arabinose from WAX [
37]. In general, the specificity and activity of arabinofuranosidases on arabinoxylans are likely mostly related to molecular details of their substrate binding sites and the presence of carbohydrate-binding modules such as CBM6, CBM35, CBM13 or CBM42, which can enable the enzymes’ attachment to the xylose polymer [
38,
39]. The CBM6 module of
AmAraf43, thus, may influence the binding ability of the enzyme to xylans, but this has not yet been experimentally demonstrated. Like
AmAraf43,
CtAbf43A from
C. thermocellum has been reported to only liberate arabinose from side chains of arabinoxylan, but not from sugar beet arabinan [
40]. Structural analysis of
CtAbf43A showed that this enzyme contained a long substrate-binding cleft that is complementary to the xylan backbone but not to an arabinan backbone [
29].
AmAraf43 from N2K1 shared 59.20% amino acid identity with
CtAbf43A, therefore its molecular determinants of substrate specificity may also apply to
AmAraf43.
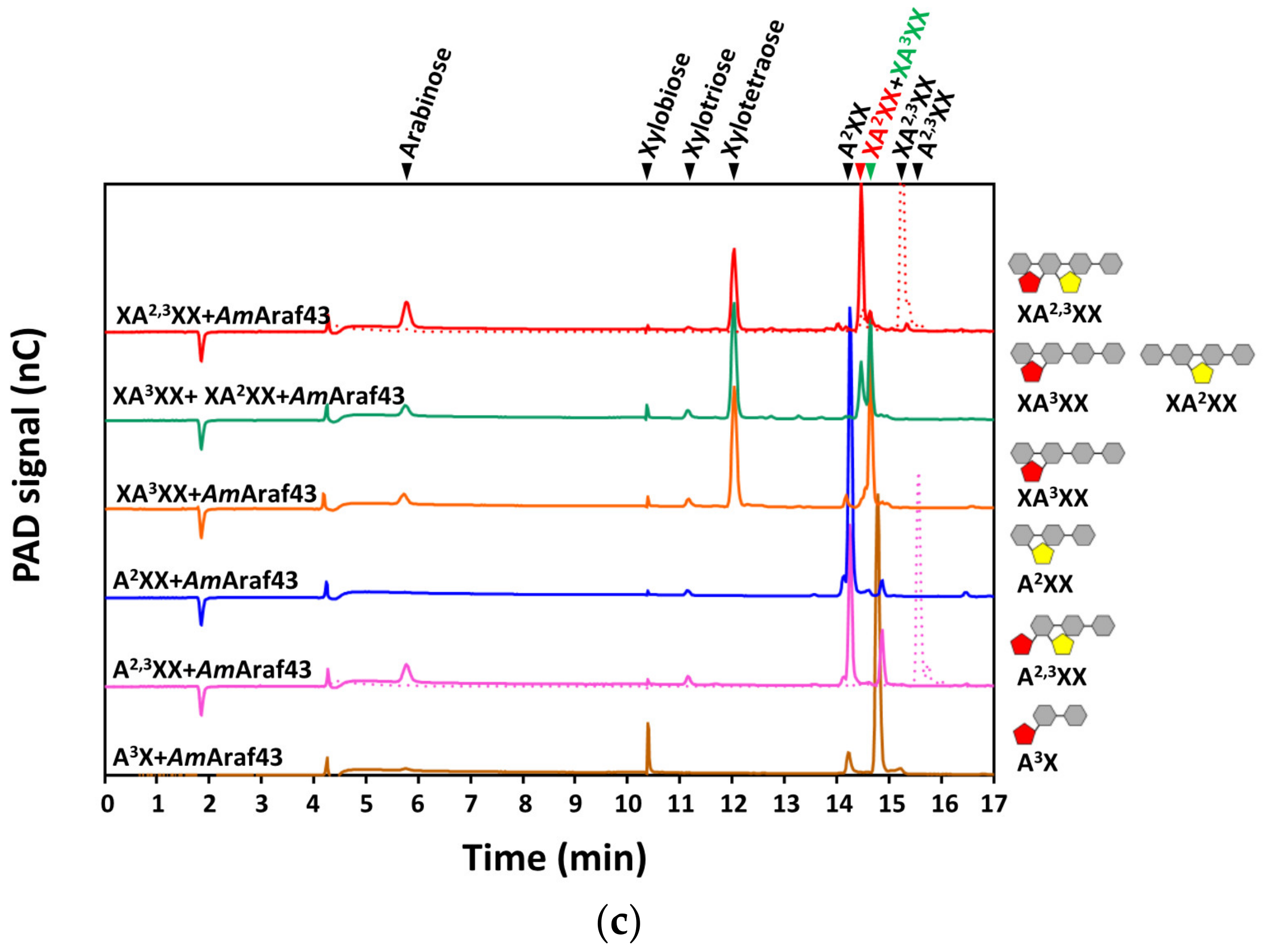
We further used defined arabinosylated arabino- and xylo-oligosaccharides to evaluate the action mode of the arabinofuranosidases of this study. Both enzymes showed cleavage activity against arabinose residues at the O-2 and/or O-3 position, but the precise nature of the side chain substitution affected the catalytic efficiency significantly. With AOS, the time required for
AmAraf51 to completely hydrolyze the substrate was negatively correlated with the complexity of the substrate oligomer (A3 > AA
3A > AA
2,3A + AA
3AA), while
AmAraf43 revealed the opposite preference (AA
2,3A +AA
3AA > AA
3A > A3), an activity towards α-1,5-arabinosyl linkages, as in A3 as a substrate, was barely detectable. Both enzymes are able to remove arabinose moieties from linear and branched XOS, but the activity of
AmAraf51 on internally twofold arabinosylated XA
2,3XX is very weak, in contrast to its high activity towards internally double substituted arabinotriose (AA
2,3A) again demonstrating this enzyme’s preference for the arabinose-based backbone. Based on this observation, the weak activity of
AmAraf51 against arabinose-substituted XOS can be classified into the arabinofuranosidase type AXHs-m,d, which has a dual activity on terminally and/or internally located monosubstituted and double substituted xylose [
21]. In comparison,
AmAraf43 has a strong preference to remove the arabinose at O-3 position in double substituted xylose residues, no matter whether this is a terminal or an internal xylose (XA
2,3XX, A
2,3XX), while single 1,3-side chain substitutions are less preferred, which matches the characteristics of AXHs-m,d3 type arabinofuranosidases [
21]. Further, with linear XOS as a substrate,
AmAraf51 also showed β-xylosidase side-activity. This is also the case with side chain modified XOS, where we found various linear XOS in the hydrolysis products. This kind of bifunctional cleavage of arabinosidic and xylosidic linkages was also found for other enzymes such as a GH51 representative from
Alicyclobacillus sp. A4, which displayed a combination of arabinofuranosidase and endoxylanase activities releasing arabinose, xylobiose, xylotriose and xylotetraose from water-soluble wheat arabinoxylan [
41]. Additionally, another GH51 arabinofuranosidase from
Paenibacillus sp. THS1 can hydrolyze not only side-chain but also main-chain glycosidic bonds in heteroxylans [
42]. It has been described that the GH51 Abf from
G. stearothermophilus T-6 can bind xylopyranosidic substrates in its active site [
43], although in this case xylanase activity was not determined. The physiological relevance of the cleavage of β-xylosidic bonds in xylans or XOS by GH51 arabinofuranosidases remains to be elucidated. In the case of
AmAraf51 of the present study, which due to the lack of a predicted N-terminal signal pepetide is postulated to be an intracellular enzyme, it is possible that both the arabinofuranosidase and the β-xylosidase activities could be of relevance during the decomposition of internalized AXOS/XOS and AOS originating from xylan and arabinan, respectively.
Enzyme thermostability is a balancing act between enzymatic activity and stability, which determines the overall efficiency over time and therefore is one of the most crucial factors that determines the temperature range of usage in industrial applications.
AmAraf51 displayed superior performance compared to
AmAraf43 in terms of overall resistance against thermoinactivation and stability at the temperature of its highest activity. The catalytic module of GH51 has a N-terminal (β/α)
8-barrel architecture followed by a β-sandwich domain with an unknown function [
1]. Enzymes with a (β/α)
8-barrel architecture from thermophiles can be stabilized by various means, such as an increased association state, additional salt bridges or hydrogen bonds [
44], leading to half-lives of GH51 enzymes ranging from hours up to several days at high temperature, for example, Abf51 from
A. clariflavus DSM 19732 had a half-life of seven days at its optimal catalyzing temperature of 60 °C [
45]. In contrast, GH43 enzymes, with a fivefold β–propeller architecture, are less studied in detail with regard to their thermostability compared with GH51 [
14].
As both
AmAraf51 and
AmAraf43 display activity with polysaccharide substrates, we finally discuss the potential of these enzymes as components of enzyme cocktails for the decomposition of plant biomass in enzyme reactors. The total hydrolysis of heteroxylan and pectic arabinan requires exoacting accessory enzymes, such as arabinofuranosidase, to generate debranched intermediates. Thereby, they are better accessible for endoacting enzymes such as endoxylanase (for arabinoxylan) and endoarabinanase (for SBA). In recent years, various studies have focused on the use of enzyme cocktails for biomass degradation. For example, Geng et al., reported that the presence of Abf51 from
Acetivibrio clariflavus (
Hungateiclostridium clariflavum) DSM 19732 [
45] dramatically improved the saccharification level of arabinoxylan (18.5 g L
−1) up to six times along with a GH11 β-1,4-xylanase (XynA) from
Thermomyces lanuginosus [
46] and a GH43 β-1,4-xylosidase (Xyl43C) from
Clostridium clariflavum [
47]. On the other hand, Bouraoui et al. showed that the combination of a GH51 arabinofuranosidase and a GH11 endoxylanase did not result in synergy [
42]. A GH11 xylanase may not be the best combination with a GH51 arabinofuranosidase because GH11 xylanases produce cleavage products with double substitutions mostly on internal rather than terminal xylopyranosyl residues [
48] (
Figure S1c), such decorations largely resist hydrolysis by GH51 and other arabinofuranosidases [
18], as was also observed by us, i.e., the catalytic efficiency of
AmAraf51 was very low with XA
2,3XX. Recently, a GH51 arabinofuranosidase (
XacAbf51) was reported to be able to cleave arabinofuranosyl linkages of internal disubstitutions of xyloparanosyl units, which can be attributed to the presence of a pocket arranged near to subsite-1 that can accommodate a second arabinofuranosyl group [
15]. In general, the hydrolytic ability of GH51 arabinofuranosidases (also including
AmAraf51) against double substitutions on terminal xylopyranosyl residues such as in A
2+3XX, suggests a superior activity on GH10- rather than GH11-xylanase-derived arabinoxylan degradation products, of which some have arabinosyl disubstitutions on terminal xylopyranosyl residues [
48]. In agreement, our WAX-RS degradation experiments (
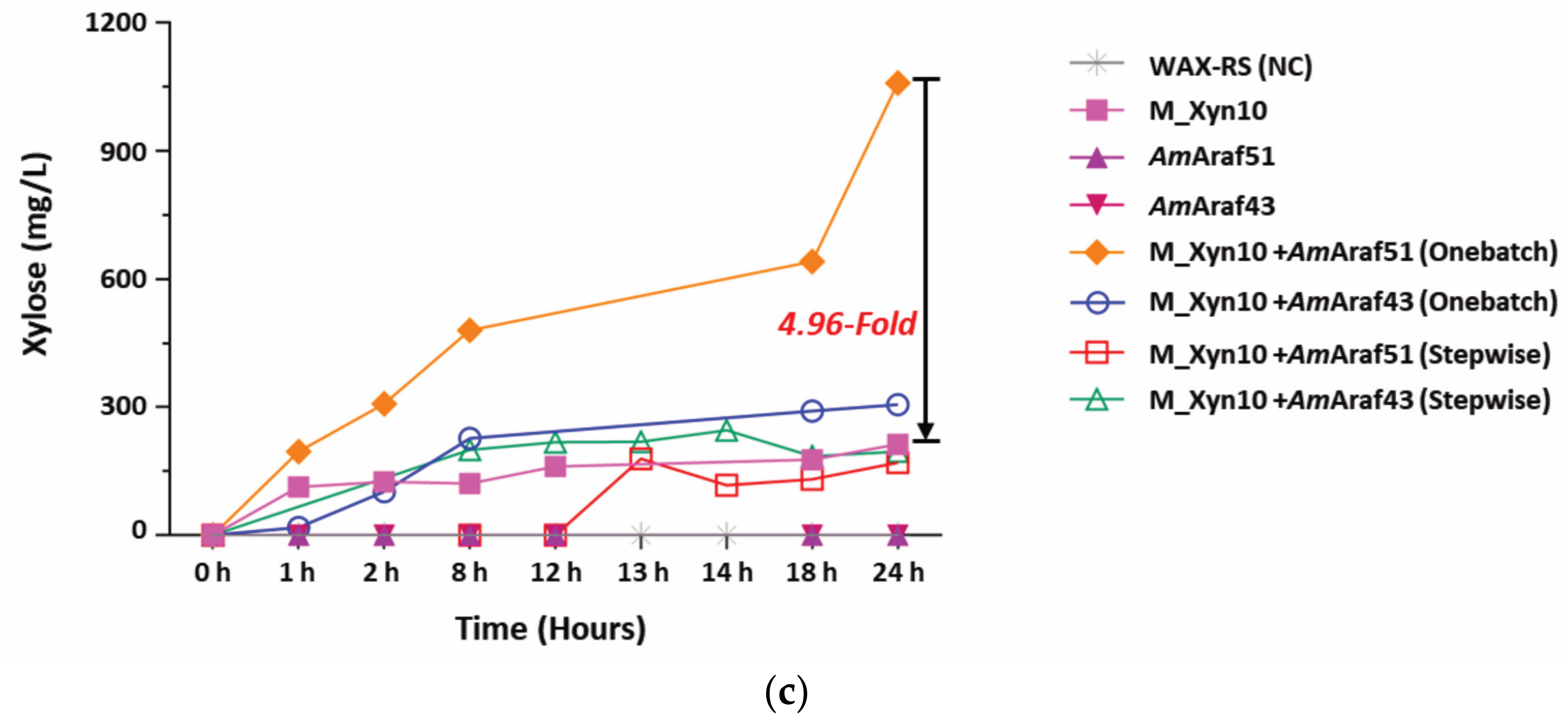
Figure 7) showed the synergistic effects of
AmAraf51 and M_Xyn10, which in combination increased the yields of arabinose almost 14-fold and of xylose almost 5-fold compared with single enzyme treatments with only
AmAraf51 or only M_Xyn10, respectively. Furthermore, the one batch treatment of SBA with
AmAraf51 and
PpAbn43 enhanced the liberation of arabinose by 3.4 fold after 6 h compared with
AmAraf51 alone, showing that
AmAraf51 may also be useful in sugar beet arabinan monomerization when combined with an endoarabinanase. Finally, a further factor thats may affect the yields of enzymatic substrate conversion is inhibition of the enzymes by cleavage products. To this end, both
AmAraf51 and
AmAraf43 compare favorably with other arabinofuranosidases (IC
50 = 100–500 mM) [
49,
50], as their
pNP-α-
l-arabinofuranoside-cleaving activity was not significantly reduced by up to 533 mM of the product arabinose (
Figure S3c).


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}