
Viruses and Type 1 Diabetes: From Enteroviruses to the Virome

 , , , and
, , , and 
Abstract
:1. Type 1 Diabetes
2. Islet Autoimmunity
3. Genetics
4. Environmental Triggers
5. Enteroviruses
6. Historical Association between EV and T1D
7. Pathogenesis Mechanisms
8. Site of Infection: Gut, Pancreas and Respiratory
9. Leaky Gut
10. Evolution of Virus Detection
11. Infant Virome
12. Virome and T1D

{kind=link}
{kind=link}
| Study; Cohort (Location); Recruitment * | Case/Control Numbers, Inclusion Criteria, Matching Strategy and Sample Numbers (Case/Control) | Sample Type/Collection; Virus Enrichment Strategy; Virus Detection Threshold | Main Findings |
|---|---|---|---|
| Lee, 2013 [238]; TEDDY (USA, Finland, Germany Sweden); High-risk HLA |
|
|
|
| Kramná, 2015 [203]; DIPP (Finland); High-risk HLA |
|
|
|
| Cinek, 2017 [240]; DIPP (Finland); High-risk HLA |
|
|
|
| Zhao, 2017 [239]; DIABIMMUNE (Finland, Estonia) High-risk HLA |
|
|
|
| Hippich, 2018 [188]; BABYDIET (Germany); High-risk HLA |
|
|
|
| Kim, 2019 [205]; VIGR (Australia); ≥1 FDR with T1D | Stool
|
|
|
Plasma
| |||
| Vehik, 2019 [150]; TEDDY (USA, Finland, Germany Sweden); High-risk HLA | IA case/control
|
|
|
T1D nested case/control
| |||
| Cinek, 2021 [243]; (Azerbaijan, Jordan, Nigeria, Sudan); New onset T1D |
|
|
|
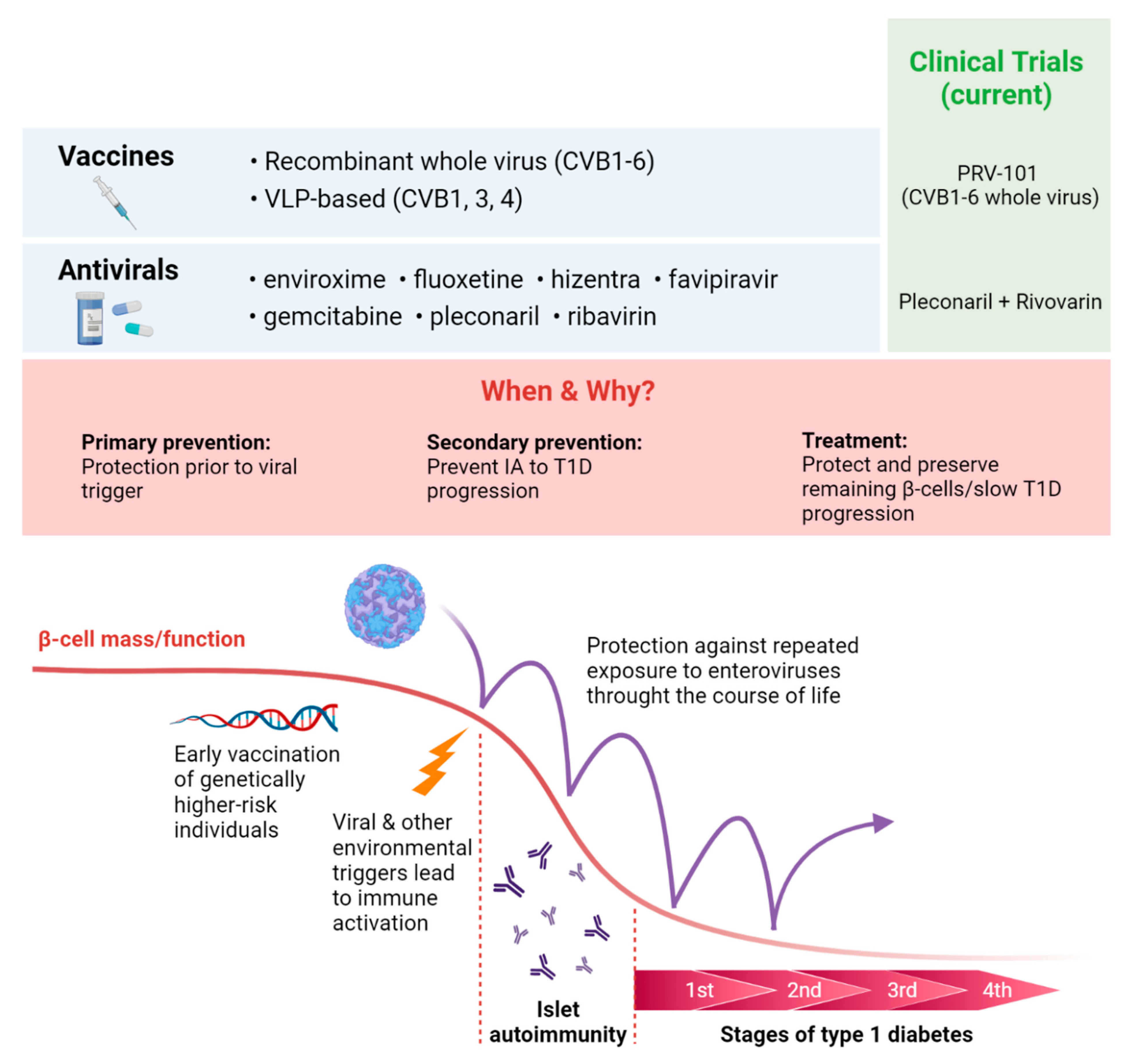
13. Antiviral Vaccines and Therapeutics for T1D Prevention
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Davis, E.J.; Kahkoska, A.R.; Jefferies, C.; Dabelea, D.; Balde, N.; Gong, C.X.; Aschner, P.; Craig, M.E. ISPAD Clinical Practice Consensus Guidelines 2018: Definition, epidemiology, and classification of diabetes in children and adolescents. Pediatr. Diabetes 2018, 19, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Atlas, 9th ed.; International Diabetes Federation (IDF): Brussles, Belgium, 2019.
- Haynes, A.; Bulsara, M.K.; Bergman, P.; Cameron, F.; Couper, J.; Craig, M.E.; Demangone, K.; Johnson, S.; Lafferty, A.; Titmuss, A.; et al. Incidence of type 1 diabetes in 0 to 14 year olds in Australia from 2002 to 2017. Pediatr. Diabetes 2020, 21, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Couper, J.J.; Haller, M.J.; Greenbaum, C.J.; Ziegler, A.-G.; Wherrett, D.K.; Knip, M.; Craig, M.E. ISPAD Clinical Practice Consensus Guidelines 2018: Stages of type 1 diabetes in children and adolescents. Pediatr. Diabetes 2018, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å. Staging Presymptomatic Type 1 Diabetes: A Scientific Statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [Green Version]
- Veijola, R.; Koskinen, M.; Helminen, O.; Hekkala, A. Dysregulation of glucose metabolism in preclinical type 1 diabetes. Pediatr. Diabetes 2016, 17, 25–30. [Google Scholar] [CrossRef]
- Borchers, A.T.; Uibo, R.; Gershwin, M.E. The geoepidemiology of type 1 diabetes. Autoimmun. Rev. 2010, 9, A355–A365. [Google Scholar] [CrossRef]
- Ferrat, L.A.; Vehik, K.; Sharp, S.A.; Lernmark, Å.; Rewers, M.J.; She, J.-X.; Ziegler, A.-G.; Toppari, J.; Akolkar, B.; Krischer, J.P.; et al. A combined risk score enhances prediction of type 1 diabetes among susceptible children. Nat. Med. 2020, 26, 1247–1255. [Google Scholar] [CrossRef]
- Taplin, C.E.; Barker, J.M. Natural Evolution, Prediction, and Prevention of Type 1 Diabetes in Youth. Endocr. Res. 2008, 33, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Cossen, K.; Muir, A. Birth Cohorts in Type 1 Diabetes: Preparing for the Payoff. J. Clin. Endocrinol. Metab. 2021, 106, e1044–e1045. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.-G.; Pflueger, M.; Winkler, C.; Achenbach, P.; Akolkar, B.; Krischer, J.P.; Bonifacio, E. Accelerated progression from islet autoimmunity to diabetes is causing the escalating incidence of type 1 diabetes in young children. J. Autoimmun. 2011, 37, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krischer, J.P.; Lynch, K.F.; Schatz, D.A.; Ilonen, J.; Lernmark, Å.; Hagopian, W.A.; Rewers, M.J.; She, J.-X.; Simell, O.G.; Toppari, J.; et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: The TEDDY study. Diabetologia 2015, 58, 980–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taplin, C.E.; Barker, J.M. Autoantibodies in type 1 diabetes. Autoimmunity 2008, 41, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Steck, A.K.; Zhang, L.; Guyer, K.M.; Jiang, L.; Armstrong, T.; Muller, S.M.; Krischer, J.; Rewers, M.; Yu, L. Electrochemiluminescence Assays for Insulin and Glutamic Acid Decarboxylase Autoantibodies Improve Prediction of Type 1 Diabetes Risk. Diabetes Technol. Ther. 2015, 17, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regnell, S.E.; Lernmark, Å. Early prediction of autoimmune (type 1) diabetes. Diabetologia 2017, 60, 1370–1381. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Zhao, Z.; Steck, A.K. T1D Autoantibodies: Room for improvement? Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 285–291. [Google Scholar] [CrossRef]
- Bauer, W.; Veijola, R.; Lempainen, J.; Kiviniemi, M.; Härkönen, T.; Toppari, J.; Knip, M.; Gyenesei, A.; Ilonen, J. Age at Seroconversion, HLA Genotype, and Specificity of Autoantibodies in Progression of Islet Autoimmunity in Childhood. J. Clin. Endocrinol. Metab. 2019, 104, 4521–4530. [Google Scholar] [CrossRef] [Green Version]
- Balke, E.M.; Balti, E.V.; Van Der Auwera, B.; Weets, I.; Costa, O.; Demeester, S.; Abrams, P.; Casteels, K.; Coeckelberghs, M.; Tenoutasse, S.; et al. Accelerated Progression to Type 1 Diabetes in the Presence ofHLA-A*24and-B*18Is Restricted to Multiple Islet Autoantibody–Positive Individuals With DistinctHLA-DQand Autoantibody Risk Profiles. Diabetes Care 2018, 41, 1076–1083. [Google Scholar] [CrossRef] [Green Version]
- Michels, A.; Zhang, L.; Khadra, A.; Kushner, J.A.; Redondo, M.J.; Pietropaolo, M. Prediction and prevention of type 1 diabetes: Update on success of prediction and struggles at prevention. Pediatr. Diabetes 2015, 16, 465–484. [Google Scholar] [CrossRef]
- Parikka, V.; Nanto-Salonen, K.; Saarinen, M.; Simell, T.; Ilonen, J.; Hyoty, H.; Veijola, R.; Knip, M.; Simell, O. Early seroconversion and rapidly increasing autoantibody concentrations predict prepubertal manifestation of type 1 diabetes in children at genetic risk. Diabetologia 2012, 55, 1926–1936. [Google Scholar] [CrossRef]
- Lönnrot, M.; Lynch, K.F.; Elding Larsson, H.; Lernmark, Å.; Rewers, M.J.; Törn, C.; Burkhardt, B.R.; Briese, T.; Hagopian, W.A.; She, J.-X.; et al. Respiratory infections are temporally associated with initiation of type 1 diabetes autoimmunity: The TEDDY study. Diabetologia 2017, 60, 1931–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosi, E.; Boulware, D.C.; Becker, D.J.; Buckner, J.H.; Geyer, S.; Gottlieb, P.A.; Henderson, C.; Kinderman, A.; Sosenko, J.M.; Steck, A.K.; et al. Impact of Age and Antibody Type on Progression From Single to Multiple Autoantibodies in Type 1 Diabetes Relatives. J. Clin. Endocrinol. Metab. 2017, 102, 2881–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knip, M.; Simell, O. Environmental triggers of type 1 diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007690. [Google Scholar] [CrossRef] [Green Version]
- Frizinsky, S.; Haj-Yahia, S.; Machnes Maayan, D.; Lifshitz, Y.; Maoz-Segal, R.; Offengenden, I.; Kidon, M.; Agmon-Levin, N. The innate immune perspective of autoimmune and autoinflammatory conditions. Rheumatology 2019, 58, vi1–vi8. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.A.; von Herrath, M.G. Potential viral pathogenic mechanism in human type 1 diabetes. Diabetologia 2014, 57, 2009–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, N.G.; Richardson, S.J. Fifty years of pancreatic islet pathology in human type 1 diabetes: Insights gained and progress made. Diabetologia 2018, 61, 2499–2506. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Ludvigsson, J.; Routray, I.; Vigård, T.; Hanås, R.; Rathsman, B.; Carlsson, A.; Särnblad, S.; Albin, A.K.; Arvidsson, C.G.; Samuelsson, U.; et al. Combined Etanercept, GAD-alum and vitamin D treatment: An open pilot trial to preserve beta cell function in recent onset type 1 diabetes. Diabetes Metab. Res. Rev. 2021, e3440. [Google Scholar] [CrossRef]
- Simmons, K.M.; Gottlieb, P.A.; Michels, A.W. Immune Intervention and Preservation of Pancreatic Beta Cell Function in Type 1 Diabetes. Curr. Diabetes Rep. 2016, 16, 97. [Google Scholar] [CrossRef]
- Eisenbarth, G.S. Prevention of Type 1A Diabetes Mellitus. Endocr. Pract. 2012, 18, 745–749. [Google Scholar] [CrossRef] [Green Version]
- Beik, P.; Ciesielska, M.; Kucza, M.; Kurczewska, A.; Kuźmińska, J.; Maćkowiak, B.; Niechciał, E. Prevention of Type 1 Diabetes: Past Experiences and Future Opportunities. J. Clin. Med. 2020, 9, 2805. [Google Scholar] [CrossRef]
- Ziegler, A.G.; Rewers, M.; Simell, O.; Simell, T.; Lempainen, J.; Steck, A.; Winkler, C.; Ilonen, J.; Veijola, R.; Knip, M.; et al. Seroconversion to Multiple Islet Autoantibodies and Risk of Progression to Diabetes in Children. JAMA 2013, 309, 2473. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.; Swensen, A.C.; Qian, W.-J. Serum biomarkers for diagnosis and prediction of type 1 diabetes. Transl. Res. 2018, 201, 13–25. [Google Scholar] [CrossRef]
- Thomson, G.; Valdes, A.M.; Noble, J.A.; Kockum, I.; Grote, M.N.; Najman, J.; Erlich, H.A.; Cucca, F.; Pugliese, A.; Steenkiste, A.; et al. Relative predispositional effects of HLA class II DRB1-DQB1 haplotypes and genotypes on type 1 diabetes: A meta-analysis. Tissue Antigens 2007, 70, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Verge, C.F.; Gianani, R.; Kawasaki, E.; Yu, L.; Pietropaolo, M.; Chase, H.P.; Eisenbarth, G.S.; Jackson, R.A. Prediction of Type I Diabetes in First-Degree Relatives Using a Combination of Insulin, GAD, and ICA512bdc/IA-2 Autoantibodies. Diabetes 1996, 45, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Redondo, M.J.; Geyer, S.; Steck, A.K.; Sharp, S.; Wentworth, J.M.; Weedon, M.N.; Antinozzi, P.; Sosenko, J.; Atkinson, M.; Pugliese, A.; et al. A Type 1 Diabetes Genetic Risk Score Predicts Progression of Islet Autoimmunity and Development of Type 1 Diabetes in Individuals at Risk. Diabetes Care 2018, 41, 1887–1894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyerlein, A.; Bonifacio, E.; Vehik, K.; Hippich, M.; Winkler, C.; Frohnert, B.I.; Steck, A.K.; Hagopian, W.A.; Krischer, J.P.; Lernmark, Å.; et al. Progression from islet autoimmunity to clinical type 1 diabetes is influenced by genetic factors: Results from the prospective TEDDY study. J. Med. Genet. 2019, 56, 602–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, S.A.; Rich, S.S.; Wood, A.R.; Jones, S.E.; Beaumont, R.N.; Harrison, J.W.; Schneider, D.A.; Locke, J.M.; Tyrrell, J.; Weedon, M.N.; et al. Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident Diagnosis. Diabetes Care 2019, 42, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Steck, A.K.; Rewers, M.J. Genetics of type 1 diabetes. Clin. Chem. 2011, 57, 176–185. [Google Scholar] [CrossRef]
- VanBuecken, D.; Lord, S.; Greenbaum, C.J. Changing the Course of Disease in Type 1 Diabetes. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Siljander, H.; Honkanen, J.; Knip, M. Microbiome and type 1 diabetes. EBioMedicine 2019, 46, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Gamble, D.R.; Kinsley, M.L.; FitzGerald, M.G.; Bolton, R.; Taylor, K.W. Viral antibodies in diabetes mellitus. Br. Med. J. 1969, 3, 627–630. [Google Scholar] [CrossRef] [Green Version]
- KimpimȧKi, T.; Kupila, A.; HȧMȧLȧInen, A.M.; Kukko, M.; Kulmala, P.; Savola, K.; Simell, T.; Keskinen, P.; Ilonen, J.; Simell, O.; et al. The First Signs of β-Cell Autoimmunity Appear in Infancy in Genetically Susceptible Children from the General Population: The Finnish Type 1 Diabetes Prediction and Prevention Study. J. Clin. Endocrinol. Metab. 2001, 86, 4782–4788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009, 52, 1143–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.W.; Kim, K.W.; Rawlinson, W.D.; Craig, M.E. Maternal virus infections in pregnancy and type 1 diabetes in their offspring: Systematic review and meta-analysis of observational studies. Rev. Med. Virol. 2018, 28, e1974. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.; Lynch, K.; Hyöty, H.; Lönnrot, M.; Driscoll, K.A.; Bennett Johnson, S. The association between stressful life events and respiratory infections during the first 4 years of life: The Environmental Determinants of Diabetes in the Young study. Stress Health 2019, 35, 289–303. [Google Scholar] [CrossRef]
- Rodriguez-Calvo, T. Enterovirus infection and type 1 diabetes: Unraveling the crime scene. Clin. Exp. Immunol. 2019, 195, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.-C.; Wang, C.-H.; Tsai, F.-J.; Hwang, K.-P.; Chen, W.; Lin, C.-C.; Li, T.-C. Enterovirus infection is associated with an increased risk of childhood type 1 diabetes in Taiwan: A nationwide population-based cohort study. Diabetologia 2015, 58, 79–86. [Google Scholar] [CrossRef]
- Simonen-Tikka, M.L.; Pflueger, M.; Klemola, P.; Savolainen-Kopra, C.; Smura, T.; Hummel, S.; Kaijalainen, S.; Nuutila, K.; Natri, O.; Roivainen, M.; et al. Human enterovirus infections in children at increased risk for type 1 diabetes: The Babydiet study. Diabetologia 2011, 54, 2995–3002. [Google Scholar] [CrossRef]
- Dahlquist, G.; Frisk, G.; Ivarsson, S.A.; Svanberg, L.; Forsgren, M.; Diderholm, H. Indications that maternal coxsackie B virus infection during pregnancy is a risk factor for childhood-onset IDDM. Diabetologia 1995, 38, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Lönnrot, M.; Salminen, K.; Knip, M.; Savola, K.; Kulmala, P.; Leinikki, P.; Hyypiä, T.; Akerblom, H.K.; Hyöty, H. Enterovirus RNA in serum is a risk factor for beta-cell autoimmunity and clinical type 1 diabetes: A prospective study. Childhood Diabetes in Finland (DiMe) Study Group. J. Med. Virol. 2000, 61, 214–220. [Google Scholar] [CrossRef]
- Viskari, H.R.; Roivainen, M.; Reunanen, A.; Pitkaniemi, J.; Sadeharju, K.; Koskela, P.; Hovi, T.; Leinikki, P.; Vilja, P.; Tuomilehto, J.; et al. Maternal First-Trimester Enterovirus Infection and Future Risk of Type 1 Diabetes in the Exposed Fetus. Diabetes 2002, 51, 2568–2571. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, C.L.; Luo, Y.X.; Isaacs, S.; Rawlinson, W.D.; Craig, M.E.; Kim, K.W. The virome in early life and childhood and development of islet autoimmunity and type 1 diabetes: A systematic review and meta-analysis of observational studies. Rev. Med. Virol. 2020, e2209. [Google Scholar] [CrossRef]
- Glanz, J.M.; Clarke, C.L.; Xu, S.; Daley, M.F.; Shoup, J.A.; Schroeder, E.B.; Lewin, B.J.; McClure, D.L.; Kharbanda, E.; Klein, N.P.; et al. Association Between Rotavirus Vaccination and Type 1 Diabetes in Children. JAMA Pediatr. 2020, 174, 455. [Google Scholar] [CrossRef]
- Honeyman, M.C.; Coulson, B.S.; Stone, N.L.; Gellert, S.A.; Goldwater, P.N.; Steele, C.E.; Couper, J.J.; Tait, B.D.; Colman, P.G.; Harrison, L.C. Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 2000, 49, 1319–1324. [Google Scholar] [CrossRef] [Green Version]
- Honeyman, M.C.; Stone, N.L.; Falk, B.A.; Nepom, G.; Harrison, L.C. Evidence for Molecular Mimicry between Human T Cell Epitopes in Rotavirus and Pancreatic Islet Autoantigens. J. Immunol. 2010, 184, 2204–2210. [Google Scholar] [CrossRef] [Green Version]
- Perrett, K.P.; Jachno, K.; Nolan, T.M.; Harrison, L.C. Association of Rotavirus Vaccination With the Incidence of Type 1 Diabetes in Children. JAMA Pediatr. 2019, 173, 280. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.L.; Sanders, N.; Tan, Y.; Allison, J.; Kay, T.W.; Coulson, B.S. Rotavirus infection accelerates type 1 diabetes in mice with established insulitis. J. Virol. 2008, 82, 6139–6149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomqvist, M.; Juhela, S.; Erkkilä, S.; Korhonen, S.; Simell, T.; Kupila, A.; Vaarala, O.; Simell, O.; Knip, M.; Ilonen, J. Rotavirus infections and development of diabetes-associated autoantibodies during the first 2 years of life. Clin. Exp. Immunol. 2002, 128, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.M.; Basu, T.; Kim, C. Lower Incidence Rate of Type 1 Diabetes after Receipt of the Rotavirus Vaccine in the United States, 2001–2017. Sci. Rep. 2019, 9, 7727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gugliesi, F.; Pasquero, S.; Griffante, G.; Scutera, S.; Albano, C.; Pacheco, S.F.C.; Riva, G.; Dell’Oste, V.; Biolatti, M. Human Cytomegalovirus and Autoimmune Diseases: Where Are We? Viruses 2021, 13, 260. [Google Scholar] [CrossRef]
- Chen, T.; Hudnall, S.D. Anatomical mapping of human herpesvirus reservoirs of infection. Mod. Pathol. 2006, 19, 726–737. [Google Scholar] [CrossRef]
- Aarnisalo, J.; Veijola, R.; Vainionpää, R.; Simell, O.; Knip, M.; Ilonen, J. Cytomegalovirus infection in early infancy: Risk of induction and progression of autoimmunity associated with type 1 diabetes. Diabetologia 2008, 51, 769–772. [Google Scholar] [CrossRef] [Green Version]
- Pak, C.; McArthur, R.; Eun, H.-M.; Yoon, J.-W. Association Of Cytomegalovirus Infection With Autoimmune Type 1 Diabetes. Lancet 1988, 332, 1–4. [Google Scholar] [CrossRef]
- Hiltunen, M.; Hyöty, H.; Karjalainen, J.; Leinikki, P.; Knip, M.; Lounamaa, R.; Kerblom, H.K. Serological evaluation of the role of cytomegalovirus in the pathogenesis of IDDM: A prospective study. Diabetologia 1995, 38, 705–710. [Google Scholar] [CrossRef]
- Ivarsson, S.A.; Lindberg, B.; Nilsson, K.O.; Ahlfors, K.; Svanberg, L. The prevalence of type 1 diabetes mellitus at follow-up of Swedish infants congenitally infected with cytomegalovirus. Diabet Med. 1993, 10, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Jenson, A.B.; Rosenberg, H.S.; Notkins, A.L. Pancreatic Islet-Cell Damage In Children With Fatal Viral Infections. Lancet 1980, 316, 354–358. [Google Scholar] [CrossRef]
- Hyöty, H.; Räasäanen, L.; Hiltunen, M.; Lehtinen, M.; Huupponen, T.; Leinikki, P. Decreased antibody reactivity to Epstein-Barr virus capsid antigen in type 1 (insulin-dependent) diabetes mellitus. APMIS 1991, 99, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Wallstrom, G.; Davis, A.; Wang, J.; Park, J.; Throop, A.; Steel, J.; Yu, X.; Wasserfall, C.; Schatz, D.; et al. Immunoproteomic Profiling of Anti-Viral Antibodies in New-Onset Type 1 Diabetes Using Protein Arrays. Diabetes 2015, 65, db150179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, A.L.; Vaziri-Sani, F.; Broberg, P.; Elfaitouri, A.; Pipkorn, R.; Blomberg, J.; Ivarsson, S.A.; Elding Larsson, H.; Lernmark, Å. Serological evaluation of possible exposure to Ljungan virus and related parechovirus in autoimmune (type 1) diabetes in children. J. Med. Virol. 2015, 87, 1130–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niklasson, B.; Heller, K.E.; Schønecker, B.; Bildsøe, M.; Daniels, T.; Hampe, C.S.; Widlund, P.; Simonson, W.T.; Schaefer, J.B.; Rutledge, E.; et al. Development of Type 1 Diabetes in Wild Bank Voles Associated With Islet Autoantibodies and the Novel Ljungan Virus. Exp. Diabesity Res. 2003, 4, 35–44. [Google Scholar] [CrossRef]
- Kolehmainen, P.; Koskiniemi, M.; Oikarinen, S.; Veijola, R.; Simell, O.; Ilonen, J.; Knip, M.; Hyöty, H.; Tauriainen, S. Human parechovirus and the risk of type 1 diabetes. J. Med. Virol. 2013, 85, 1619–1623. [Google Scholar] [CrossRef]
- Ruiz, P.L.D.; Tapia, G.; Bakken, I.J.; Håberg, S.E.; Hungnes, O.; Gulseth, H.L.; Stene, L.C. Pandemic influenza and subsequent risk of type 1 diabetes: A nationwide cohort study. Diabetologia 2018, 61, 1996–2004. [Google Scholar] [CrossRef] [Green Version]
- Piccini, B.; Toni, S.; Lenzi, L.; Guasti, M.; Barm, F.; De Martino, M. Type 1 Diabetes Onset and Pandemic Influenza A (H1N1). Int. J. Immunopathol. Pharmacol. 2012, 25, 547–549. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, K.; Salimi, V.; Rezaei, F.; Jalilian, F.A.; Ghavami, N.; Azad, T.M. Potential of H1N1 influenza A virus as an air borne pathogen to induce infectivity in pancreas: A mouse model study. J. Environ. Health Sci. Eng. 2020, 18, 303–310. [Google Scholar] [CrossRef]
- Kasuga, A. Insulin-Dependent Diabetes Mellitus Associated with Parvovirus B19 Infection. Ann. Intern. Med. 1996, 125, 700. [Google Scholar] [CrossRef]
- Munakata, Y.; Kodera, T.; Saito, T.; Sasaki, T. Rheumatoid arthritis, type 1 diabetes, and Graves’ disease after acute parvovirus B19 infection. Lancet 2005, 366, 780. [Google Scholar] [CrossRef]
- Hyoty, H.; Leinikki, P.; Reunanen, A.; Ilonen, J.; Surcel, H.M.; Rilva, A.; Kaar, M.L.; Huupponen, T.; Hakulinen, A.; Makela, A.L.; et al. Mumps infections in the etiology of type 1 (insulin-dependent) diabetes. Diabetes Res. 1988, 9, 111–116. [Google Scholar]
- Ramondetti, F.; Sacco, S.; Comelli, M.; Bruno, G.; Falorni, A.; Iannilli, A.; D’Annunzio, G.; Iafusco, D.; Songini, M.; Toni, S.; et al. Type 1 diabetes and measles, mumps and rubella childhood infections within the Italian Insulin-dependent Diabetes Registry. Diabet. Med. 2012, 29, 761–766. [Google Scholar] [CrossRef]
- Hyöty, H.; Hiltunen, M.; Reunanen, A.; Leinikki, P.; Vesikari, T.; Lounamaa, R.; Tuomilehto, J.; Akerblom, H.K. Decline of mumps antibodies in type 1 (insulin-dependent) diabetic children and a plateau in the rising incidence of type 1 diabetes after introduction of the mumps-measles-rubella vaccine in Finland. Childhood Diabetes in Finland Study Group. Diabetologia 1993, 36, 1303–1308. [Google Scholar] [CrossRef] [Green Version]
- Gale, E.A.M. Congenital rubella: Citation virus or viral cause of type 1 diabetes? Diabetologia 2008, 51, 1559–1566. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg-Fellner, F.; Witt, M.E.; Yagihashi, S.; Dobersen, M.J.; Taub, F.; Fedun, B.; McEvoy, R.C.; Roman, S.H.; Davies, T.F.; Cooper, L.Z.; et al. Congenital rubella syndrome as a model for Type 1 (insulin-dependent) diabetes mellitus: Increased prevalence of islet cell surface antibodies. Diabetologia 1984, 27, 87–89. [Google Scholar] [CrossRef]
- Rayfield, E.J. Effects of rubella virus infection on islet function. Curr. Top. Microbiol. Immunol. 1990, 156, 63–74. [Google Scholar] [CrossRef]
- Menser, M.; Dods, L.; Harley, J.D. A Twenty-Five-Year Follow-Up Of Congenital Rubella. Lancet 1967, 290, 1347–1350. [Google Scholar] [CrossRef]
- Conrad, B.; Weissmahr, R.N.; Böni, J.; Arcari, R.; Schüpbach, J.; Mach, B. A human endogenous retroviral superantigen as candidate autoimmune gene in type I diabetes. Cell 1997, 90, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef] [PubMed]
- Dechaumes, A.; Bertin, A.; Sane, F.; Levet, S.; Varghese, J.; Charvet, B.; Gmyr, V.; Kerr-Conte, J.; Pierquin, J.; Arunkumar, G.; et al. Coxsackievirus-B4 Infection Can Induce the Expression of Human Endogenous Retrovirus W in Primary Cells. Microorganisms 2020, 8, 1335. [Google Scholar] [CrossRef] [PubMed]
- Levet, S.; Charvet, B.; Bertin, A.; Deschaumes, A.; Perron, H.; Hober, D. Human Endogenous Retroviruses and Type 1 Diabetes. Curr. Diabetes Rep. 2019, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, W.C.; Rawlinson, W.D.; Craig, M.E. Enterovirus infection and type 1 diabetes mellitus: Systematic review and meta-analysis of observational molecular studies. BMJ 2011, 342, d35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanter, M.; Sork, H.; Tuomela, S.; Flodström-Tullberg, M. Genetic and Environmental Interaction in Type 1 Diabetes: A Relationship Between Genetic Risk Alleles and Molecular Traits of Enterovirus Infection? Curr. Diabetes Rep. 2019, 19, 82. [Google Scholar] [CrossRef] [Green Version]
- Cinek, O.; Tapia, G.; Witsø, E.; Kramna, L.; Holkova, K.; Rasmussen, T.; Stene, L.C.; Rønningen, K.S. Enterovirus RNA in Peripheral Blood May Be Associated with the Variants of rs1990760, a Common Type 1 Diabetes Associated Polymorphism in IFIH1. PLoS ONE 2012, 7, e48409. [Google Scholar] [CrossRef] [Green Version]
- Pang, L.; Gong, X.; Liu, N.; Xie, G.; Gao, W.; Kong, G.; Li, X.; Zhang, J.; Jin, Y.; Duan, Z. A polymorphism in melanoma differentiation-associated gene 5 may be a risk factor for enterovirus 71 infection. Clin. Microbiol. Infect. 2014, 20, O711–O717. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shaked, I.; Stanford, S.M.; Zhou, W.; Curtsinger, J.M.; Mikulski, Z.; Shaheen, Z.R.; Cheng, G.; Sawatzke, K.; Campbell, A.M. The Autoimmunity-Associated Gene PTPN22 Potentiates Toll-like Receptor-Driven, Type 1 Interferon-Dependent Immunity. Immunity 2013, 39, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabtree, J.N.; He, W.; Guan, W.; Flage, M.; Miller, M.S.; Peterson, E.J. Autoimmune VariantPTPN22C1858T Is Associated With Impaired Responses to Influenza Vaccination. J. Infect. Dis. 2016, 214, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Craig, M.E.; Kim, K.W.; Isaacs, S.R.; Penno, M.A.; Hamilton-Williams, E.E.; Couper, J.J.; Rawlinson, W.D. Early-life factors contributing to type 1 diabetes. Diabetologia 2019, 62, 1823–1834. [Google Scholar] [CrossRef] [Green Version]
- Penno, M.A.; Couper, J.J.; Craig, M.E.; Colman, P.G.; Rawlinson, W.D.; Cotterill, A.M.; Jones, T.W.; Harrison, L.C. Environmental determinants of islet autoimmunity (ENDIA): A pregnancy to early life cohort study in children at-risk of type 1 diabetes. BMC Pediatr. 2013, 13, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenfeld, E.; Domingo, E.; Roos, R.P. The Picornaviruses; ASM Press: Washington, DC, USA, 2010. [Google Scholar]
- Sells, C.J.; Carpenter, R.L.; Ray, C.G. Sequelae of Central-Nervous-System Enterovirus Infections. N. Engl. J. Med. 1975, 293, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Onkamo, P.; Vaananen, S.; Karvonen, M.; Tuomilehto, J. Worldwide increase in incidence of Type I diabetes—The analysis of the data on published incidence trends. Diabetologia 1999, 42, 1395–1403. [Google Scholar] [CrossRef] [Green Version]
- Bopegamage, S.; Kovacova, J.; Vargova, A.; Motusova, J.; Petrovicova, A.; Benkovicova, M.; Gomolcak, P.; Bakkers, J.; Van Kuppeveld, F.; Melchers, W.J.G.; et al. Coxsackie B virus infection of mice: Inoculation by the oral route protects the pancreas from damage, but not from infection. J. Gen. Virol. 2005, 86, 3271–3280. [Google Scholar] [CrossRef]
- Craig, M.E.; Nair, S.; Stein, H.; Rawlinson, W.D. Viruses and type 1 diabetes: A new look at an old story. Pediatr. Diabetes 2013, 14, 149–158. [Google Scholar] [CrossRef]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Palmenberg, A.C.; Gern, J.E. Classification and evolution of human rhinoviruses. Methods Mol. Biol. 2015, 1221, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hober, D.; Sauter, P. Pathogenesis of type 1 diabetes mellitus: Interplay between enterovirus and host. Nat. Rev. Endocrinol. 2010, 6, 279–289. [Google Scholar] [CrossRef]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Petzold, A.; Solimena, M.; Knoch, K.-P. Mechanisms of Beta Cell Dysfunction Associated With Viral Infection. Curr. Diabetes Rep. 2015, 15, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Wang, Q.; Sintchenko, V.; Gilbert, G.L.; O’Sullivan, M.V.; Iredell, J.R.; Dwyer, D.E. Use of the 5’ untranslated region and VP1 region to examine the molecular diversity in enterovirus B species. J. Med. Microbiol 2014, 63, 1339–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, P.; Kammerer, U.; Korn, K.; Mulders, M.N.; Poyry, T.; Weissbrich, B.; Kandolf, R.; Cleator, G.M.; van Loon, A.M. Molecular typing of enteroviruses: Current status and future requirements. The European Union Concerted Action on Virus Meningitis and Encephalitis. Clin. Microbiol. Rev. 1998, 11, 202–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, M.E.; Robertson, P.; Howard, N.J.; Silink, M.; Rawlinson, W.D. Diagnosis of enterovirus infection by genus-specific PCR and enzyme-linked immunosorbent assays. J. Clin. Microbiol. 2003, 41, 841–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ylipaasto, P.; Klingel, K.; Lindberg, A.M.; Otonkoski, T.; Kandolf, R.; Hovi, T.; Roivainen, M. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia 2004, 47, 225–239. [Google Scholar] [CrossRef] [Green Version]
- Ifie, E.; Russell, M.A.; Dhayal, S.; Leete, P.; Sebastiani, G.; Nigi, L.; Dotta, F.; Marjomäki, V.; Eizirik, D.L.; Morgan, N.G.; et al. Unexpected subcellular distribution of a specific isoform of the Coxsackie and adenovirus receptor, CAR-SIV, in human pancreatic beta cells. Diabetologia 2018, 61, 2344–2355. [Google Scholar] [CrossRef] [Green Version]
- Hodik, M.; Skog, O.; Lukinius, A.; Isaza-Correa, J.M.; Kuipers, J.; Giepmans, B.N.G.; Frisk, G. Enterovirus infection of human islets of Langerhans affects β-cell function resulting in disintegrated islets, decreased glucose stimulated insulin secretion and loss of Golgi structure. BMJ Open Diabetes Res. Care 2016, 4, e000179. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.J.; Morgan, N.G. Enteroviral infections in the pathogenesis of type 1 diabetes: New insights for therapeutic intervention. Curr. Opin. Pharm. 2018, 43, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.F. The seasonal variation in the onset of acute diabetes: The age and sex factors in 1,000 diabetic patients. Arch. Intern. Med. 1926, 37, 861. [Google Scholar] [CrossRef]
- Gundersen, E. Is Diabetes of Infectious Origin? J. Infect. Dis. 1927, 41, 197–202. [Google Scholar] [CrossRef]
- Gamble, D.R.; Taylor, K.W. Seasonal incidence of diabetes mellitus. Br. Med. J. 1969, 3, 631–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moltchanova, E.V.; Schreier, N.; Lammi, N.; Karvonen, M. Seasonal variation of diagnosis of Type 1 diabetes mellitus in children worldwide. Diabet. Med. 2009, 26, 673–678. [Google Scholar] [CrossRef]
- Szypowska, A.; Ramotowska, A.; Wysocka-Mincewicz, M.; Mazur, A.; Lisowicz, L.; Beń-Skowronek, I.; Sieniawska, J.; Klonowska, B.; Charemska, D.; Nawrotek, J.; et al. Seasonal Variation in Month of Diagnosis of Polish Children with Type 1 Diabetes—A Multicenter Study. Exp. Clin. Endocrinol. Diabetes 2019, 127, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Spaans, E.A.; van Dijk, P.R.; Groenier, K.H.; Brand, P.L.; Reeser, M.H.; Bilo, H.J.; Kleefstra, N. Seasonality of diagnosis of type 1 diabetes mellitus in the Netherlands (Young Dudes-2). J. Pediatr. Endocrinol. Metab. 2016, 29, 657–661. [Google Scholar] [CrossRef] [Green Version]
- Watad, A.; Azrielant, S.; Bragazzi, N.L.; Sharif, K.; David, P.; Katz, I.; Aljadeff, G.; Quaresma, M.; Tanay, G.; Adawi, M.; et al. Seasonality and autoimmune diseases: The contribution of the four seasons to the mosaic of autoimmunity. J. Autoimmun. 2017, 82, 13–30. [Google Scholar] [CrossRef]
- Geravandi, S.; Liu, H.; Maedler, K. Enteroviruses and T1D: Is It the Virus, the Genes or Both Which Cause T1D. Microorganisms 2020, 8, 1017. [Google Scholar] [CrossRef]
- Knip, M.; Veijola, R.; Virtanen, S.M.; Hyoty, H.; Vaarala, O.; Akerblom, H.K. Environmental triggers and determinants of type 1 diabetes. Diabetes 2005, 54 (Suppl. 2), S125–S136. [Google Scholar] [CrossRef] [Green Version]
- Nair, S. Mechanisms of Enterovirus Induced β-Cells Destruction: Role of Cytokines and Signalling Pathways; University of New South Wales: Sydney, NSW, Australia, 2014; Unpublished work. [Google Scholar]
- Sioofy-Khojine, A.-B.; Oikarinen, S.; Honkanen, H.; Huhtala, H.; Lehtonen, J.P.; Briese, T.; Hyöty, H. Molecular epidemiology of enteroviruses in young children at increased risk of type 1 diabetes. PLoS ONE 2018, 13, e0201959. [Google Scholar] [CrossRef] [Green Version]
- Rasilainen, S.; Ylipaasto, P.; Roivainen, M.; Lapatto, R.; Hovi, T.; Otonkoski, T. Mechanisms of coxsackievirus B5 mediated beta-cell death depend on the multiplicity of infection. J. Med. Virol. 2004, 72, 586–596. [Google Scholar] [CrossRef]
- Kanno, T.; Kim, K.; Kono, K.; Drescher, K.M.; Chapman, N.M.; Tracy, S. Group B coxsackievirus diabetogenic phenotype correlates with replication efficiency. J. Virol. 2006, 80, 5637–5643. [Google Scholar] [CrossRef] [Green Version]
- Krogvold, L.; Edwin, B.; Buanes, T.; Frisk, G.; Skog, O.; Anagandula, M.; Korsgren, O.; Undlien, D.; Eike, M.C.; Richardson, S.J.; et al. Detection of a Low-Grade Enteroviral Infection in the Islets of Langerhans of Living Patients Newly Diagnosed With Type 1 Diabetes. Diabetes 2015, 64, 1682–1687. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.J.; Rodriguez-Calvo, T.; Gerling, I.C.; Mathews, C.E.; Kaddis, J.S.; Russell, M.A.; Zeissler, M.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; et al. Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 2016, 59, 2448–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, M.; Krogvold, L.; Kuric, E.; Dahl-Jørgensen, K.; Skog, O. Expression of Interferon-Stimulated Genes in Insulitic Pancreatic Islets of Patients Recently Diagnosed With Type 1 Diabetes. Diabetes 2016, 65, 3104–3110. [Google Scholar] [CrossRef] [Green Version]
- Kuric, E.; Seiron, P.; Krogvold, L.; Edwin, B.; Buanes, T.; Hanssen, K.F.; Skog, O.; Dahl-Jørgensen, K.; Korsgren, O. Demonstration of Tissue Resident Memory CD8 T Cells in Insulitic Lesions in Adult Patients with Recent-Onset Type 1 Diabetes. Am. J. Pathol. 2017, 187, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Oikarinen, M.; Laiho, J.E.; Oikarinen, S.; Richardson, S.J.; Kusmartseva, I.; Campbell-Thompson, M.; Morgan, N.G.; Pugliese, A.; Tauriainen, S.; Toniolo, A.; et al. Detection of enterovirus protein and RNA in multiple tissues from nPOD organ donors with type 1 diabetes. bioRxiv 2018, 459347. [Google Scholar] [CrossRef]
- Apaolaza, P.S.; Balcacean, D.; Zapardiel-Gonzalo, J.; Nelson, G.; Lenchik, N.; Akhbari, P.; Gerling, I.; Richardson, S.J.; Rodriguez-Calvo, T. Islet expression of type I interferon response sensors is associated with immune infiltration and viral infection in type 1 diabetes. Sci. Adv. 2021, 7, eabd6527. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.M.; Genoni, A.; Broccolo, F.; Denaro, M.; Pugliese, A.; Basolo, F.; Toniolo, A. Immune Transcriptome of Cells Infected with Enterovirus Strains Obtained from Cases of Type 1 Diabetes. Microorganisms 2020, 8, 1031. [Google Scholar] [CrossRef]
- Oikarinen, S.; Martiskainen, M.; Tauriainen, S.; Huhtala, H.; Ilonen, J.; Veijola, R.; Simell, O.; Knip, M.; Hyöty, H. Enterovirus RNA in Blood Is Linked to the Development of Type 1 Diabetes. Diabetes 2011, 60, 276–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honkanen, H.; Oikarinen, S.; Nurminen, N.; Laitinen, O.H.; Huhtala, H.; Lehtonen, J.; Ruokoranta, T.; Hankaniemi, M.M.; Lecouturier, V.; Almond, J.W.; et al. Detection of enteroviruses in stools precedes islet autoimmunity by several months: Possible evidence for slowly operating mechanisms in virus-induced autoimmunity. Diabetologia 2017, 60, 424–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Op De Beeck, A.; Eizirik, D.L. Viral infections in type 1 diabetes mellitus—Why the β cells? Nat. Rev. Endocrinol. 2016, 12, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, M.S.; Bradley, L.M.; Harbertson, J.; Krahl, T.; Lee, J.; Sarvennick, N. Diabetes induced by Coxsackie virus: Initiation by bystander damage and not molecular mimicry. Nat. Med. 1998, 4, 781–785. [Google Scholar] [CrossRef]
- Pane, J.A.; Coulson, B.S. Lessons from the mouse: Potential contribution of bystander lymphocyte activation by viruses to human type 1 diabetes. Diabetologia 2015, 58, 1149–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, S.I.; Tse, H.M. Innate Viral Sensor MDA5 and Coxsackievirus Interplay in Type 1 Diabetes Development. Microorganisms 2020, 8, 993. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef]
- Kobayashi, T.; Tanaka, S.; Aida, K. Unique pathological changes in the pancreas of fulminant type 1 diabetes. Diabetol. Int. 2020, 11, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Galleri, L.; Sebastiani, G.; Vendrame, F.; Grieco, F.A.; Spagnuolo, I.; Dotta, F. Viral infections and diabetes. Adv. Exp. Med. Biol. 2012, 771, 252–271. [Google Scholar] [CrossRef]
- Von Herrath, M.; Sanda, S.; Herold, K. Type 1 diabetes as a relapsing-remitting disease? Nat. Rev. Immunol. 2007, 7, 988–994. [Google Scholar] [CrossRef]
- Thomas, H.E.; Graham, K.L.; Chee, J.; Thomas, R.; Kay, T.W.; Krishnamurthy, B. Proinflammatory cytokines contribute to development and function of regulatory T cells in type 1 diabetes. Ann. N. Y. Acad. Sci. 2013, 1283, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Netanyah, E.; Calafatti, M.; Arvastsson, J.; Cabrera-Rode, E.; Cilio, C.M.; Sarmiento, L. Extracellular Vesicles Released by Enterovirus-Infected EndoC-βH1 Cells Mediate Non-Lytic Viral Spread. Microorganisms 2020, 8, 1753. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Perez-Serna, A.A.; Babiloni-Chust, I.; Dos Santos, R.S. Type I interferons as key players in pancreatic β-cell dysfunction in type 1 diabetes. Int. Rev. Cell Mol. Biol. 2021, 359, 1–80. [Google Scholar] [CrossRef] [PubMed]
- Akhbari, P.; Richardson, S.J.; Morgan, N.G. Type 1 Diabetes: Interferons and the Aftermath of Pancreatic Beta-Cell Enteroviral Infection. Microorganisms 2020, 8, 1419. [Google Scholar] [CrossRef]
- Chehadeh, W.; Kerr-Conte, J.; Pattou, F.; Alm, G.; Lefebvre, J.; Wattré, P.; Hober, D. Persistent Infection of Human Pancreatic Islets by Coxsackievirus B Is Associated with Alpha Interferon Synthesis in β Cells. J. Virol. 2000, 74, 10153–10164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vehik, K.; Lynch, K.F.; Wong, M.C.; Tian, X.; Ross, M.C.; Gibbs, R.A.; Ajami, N.J.; Petrosino, J.F.; Rewers, M.; Toppari, J.; et al. Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med. 2019, 25, 1865–1872. [Google Scholar] [CrossRef]
- Nekoua, M.P.; Dechaumes, A.; Sane, F.; Alidjinou, E.K.; Moutairou, K.; Yessoufou, A.; Hober, D. Enteroviral Pathogenesis of Type 1 Diabetes: The Role of Natural Killer Cells. Microorganisms 2020, 8, 989. [Google Scholar] [CrossRef]
- Nekoua, M.P.; Bertin, A.; Sane, F.; Alidjinou, E.K.; Lobert, D.; Trauet, J.; Hober, C.; Engelmann, I.; Moutairou, K.; Yessoufou, A.; et al. Pancreatic beta cells persistently infected with coxsackievirus B4 are targets of NK cell-mediated cytolytic activity. Cell. Mol. Life Sci. 2020, 77, 179–194. [Google Scholar] [CrossRef]
- Oikarinen, M.; Tauriainen, S.; Oikarinen, S.; Honkanen, T.; Collin, P.; Rantala, I.; Maki, M.; Kaukinen, K.; Hyoty, H. Type 1 Diabetes Is Associated With Enterovirus Infection in Gut Mucosa. Diabetes 2012, 61, 687–691. [Google Scholar] [CrossRef] [Green Version]
- Ylipaasto, P.; Smura, T.; Gopalacharyulu, P.; Paananen, A.; Seppänen-Laakso, T.; Kaijalainen, S.; Ahlfors, H.; Korsgren, O.; Lakey, J.R.T.; Lahesmaa, R.; et al. Enterovirus-induced gene expression profile is critical for human pancreatic islet destruction. Diabetologia 2012, 55, 3273–3283. [Google Scholar] [CrossRef] [Green Version]
- Harrath, R.; Bourlet, T.; Delézay, O.; Douche-Aourik, F.; Omar, S.; Aouni, M.; Pozzetto, B. Coxsackievirus B3 replication and persistence in intestinal cells from mice infected orally and in the human CaCo-2 cell line. J. Med. Virol. 2004, 74, 283–290. [Google Scholar] [CrossRef]
- Ferrer-Orta, C.; Ferrero, D.; Verdaguer, N. RNA-Dependent RNA Polymerases of Picornaviruses: From the Structure to Regulatory Mechanisms. Viruses 2015, 7, 4438–4460. [Google Scholar] [CrossRef] [Green Version]
- Campagnola, G.; McDonald, S.; Beaucourt, S.; Vignuzzi, M.; Peersen, O.B. Structure-function relationships underlying the replication fidelity of viral RNA-dependent RNA polymerases. J. Virol. 2015, 89, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, A.M.; Andersson, P.; Savolainen, C.; Mulders, M.N.; Hovi, T. Evolution of the genome of Human enterovirus B: Incongruence between phylogenies of the VP1 and 3CD regions indicates frequent recombination within the species. J. Gen. Virol. 2003, 84, 1223–1235. [Google Scholar] [CrossRef]
- Honkimaa, A.; Kimura, B.; Sioofy-Khojine, A.B.; Lin, J.; Laiho, J.; Oikarinen, S.; Hyöty, H. Genetic Adaptation of Coxsackievirus B1 during Persistent Infection in Pancreatic Cells. Microorganisms 2020, 8, 1790. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Tracy, S.; Tapprich, W.; Bailey, J.; Lee, C.K.; Kim, K.; Barry, W.H.; Chapman, N.M. 5’-Terminal deletions occur in coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative-strand viral RNA. J. Virol. 2005, 79, 7024–7041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tracy, S.; Smithee, S.; Alhazmi, A.; Chapman, N. Coxsackievirus can persist in murine pancreas by deletion of 5′ terminal genomic sequences. J. Med. Virol. 2015, 87, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Chapman, N.M.; Kim, K.S.; Drescher, K.M.; Oka, K.; Tracy, S. 5’ terminal deletions in the genome of a coxsackievirus B2 strain occurred naturally in human heart. Virology 2008, 375, 480–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunziker, I.P.; Cornell, C.T.; Whitton, J.L. Deletions within the 5′ UTR of coxsackievirus B3: Consequences for virus translation and replication. Virology 2007, 360, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Glenet, M.; Heng, L.; Callon, D.; Lebreil, A.-L.; Gretteau, P.-A.; Nguyen, Y.; Berri, F.; Andreoletti, L. Structures and Functions of Viral 5′ Non-Coding Genomic RNA Domain-I in Group-B Enterovirus Infections. Viruses 2020, 12, 919. [Google Scholar] [CrossRef]
- Paananen, A.; Ylipaasto, P.; Smura, T.; Lempinen, M.; Galama, J.; Roivainen, M. A single amino acid substitution in viral VP1 protein alters the lytic potential of clone-derived variants of echovirus 9 DM strain in human pancreatic islets. J. Med. Virol. 2013, 85, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Caine, E.A.; Moncla, L.H.; Ronderos, M.D.; Friedrich, T.C.; Osorio, J.E. A Single Mutation in the VP1 of Enterovirus 71 Is Responsible for Increased Virulence and Neurotropism in Adult Interferon-Deficient Mice. J. Virol. 2016, 90, 8592–8604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseligka, E.D.; Sobo, K.; Stoppini, L.; Cagno, V.; Abdul, F.; Piuz, I.; Meylan, P.; Huang, S.; Constant, S.; Tapparel, C. A VP1 mutation acquired during an enterovirus 71 disseminated infection confers heparan sulfate binding ability and modulates ex vivo tropism. PLoS Pathog. 2018, 14, e1007190. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, J.; Yao, M.X.; Zhang, Y.W.; Hu, T.; Carr, M.J.; Duchêne, S.; Zhang, X.C.; Zhang, Z.J.; Zhou, H.; et al. Genome Analysis of Coxsackievirus A4 Isolates From Hand, Foot, and Mouth Disease Cases in Shandong, China. Front. Microbiol. 2019, 10, 1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Blake, N.W.; Ouyang, X.; Pandolfino, Y.A.; Morgan-Capner, P.; Archard, L.C. A single amino acid substitution in the capsid protein VP1 of Coxsackievirus B3 (CVB3) alters plaque phenotype in Vero cells but not cardiovirulence in a mouse model. Arch. Virol. 1995, 140, 959–966. [Google Scholar] [CrossRef]
- Halim, S.; Ramsingh, A.I. A point mutation in VP1 of coxsackievirus B4 alters antigenicity. Virology 2000, 269, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaacs, S.R.; Wang, J.; Kim, K.W.; Yin, C.; Zhou, L.; Mi, Q.S.; Craig, M.E. MicroRNAs in Type 1 Diabetes: Complex Interregulation of the Immune System, beta Cell Function and Viral Infections. Curr. Diabetes Rep. 2016, 16, 133. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, I.; Alidjinou, E.K.; Bertin, A.; Bossu, J.; Villenet, C.; Figeac, M.; Sane, F.; Hober, D. Persistent coxsackievirus B4 infection induces microRNA dysregulation in human pancreatic cells. Cell. Mol. Life Sci. 2017, 74, 3851–3861. [Google Scholar] [CrossRef]
- Engelmann, I.; Alidjinou, E.K.; Bertin, A.; Sane, F.; Hober, D. miRNAs in enterovirus infection. Crit. Rev. Microbiol. 2018, 44, 701–714. [Google Scholar] [CrossRef]
- Kim, K.W.; Ho, A.; Alshabee-Akil, A.; Hardikar, A.A.; Kay, T.W.H.; Rawlinson, W.D.; Craig, M.E. Coxsackievirus B5 Infection Induces Dysregulation of microRNAs Predicted to Target Known Type 1 Diabetes Risk Genes in Human Pancreatic Islets. Diabetes 2016, 65, 996–1003. [Google Scholar] [CrossRef] [Green Version]
- Eringsmark Regnéll, S.; Lernmark, Å. The environment and the origins of islet autoimmunity and Type 1 diabetes. Diabet. Med. 2013, 30, 155–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Ylipaasto, P.; Kutlu, B.; Rasilainen, S.; Rasschaert, J.; Salmela, K.; Teerijoki, H.; Korsgren, O.; Lahesmaa, R.; Hovi, T.; Eizirik, D.L.; et al. Global profiling of coxsackievirus- and cytokine-induced gene expression in human pancreatic islets. Diabetologia 2005, 48, 1510–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, V.M.; Ringqvist, E.E.; Larsson, P.G.; Domsgen, E.; Holmlund, U.; Sverremark-Ekström, E.; Flodström-Tullberg, M. Inhibition of Type III Interferon Expression in Intestinal Epithelial Cells—A Strategy Used by Coxsackie B Virus to Evade the Host’s Innate Immune Response at the Primary Site of Infection? Microorganisms 2021, 9, 105. [Google Scholar] [CrossRef]
- Tauriainen, S.; Oikarinen, S.; Oikarinen, M.; Hyoty, H. Enteroviruses in the pathogenesis of type 1 diabetes. Semin. Immunopathol. 2011, 33, 45–55. [Google Scholar] [CrossRef]
- Royston, L.; Tapparel, C. Rhinoviruses and Respiratory Enteroviruses: Not as Simple as ABC. Viruses 2016, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Walker, G.J.; Stelzer-Braid, S.; Shorter, C.; Honeywill, C.; Wynn, M.; Willenborg, C.; Barnes, P.; Kang, J.; Pierse, N.; Crane, J.; et al. Viruses associated with acute respiratory infection in a community-based cohort of healthy New Zealand children. J. Med. Virol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Ponsonby, A.-L.; Pezic, A.; Cochrane, J.; Cameron, F.J.; Pascoe, M.; Kemp, A.; Dwyer, T. Infant anthropometry, early life infection, and subsequent risk of type 1 diabetes mellitus: A prospective birth cohort study. Pediatr. Diabetes 2011, 12, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.; Witso, E.; Tapia, G.; Stene, L.C.; Ronningen, K.S. Self-reported lower respiratory tract infections and development of islet autoimmunity in children with the type 1 diabetes high-risk HLA genotype: The MIDIA study. Diabetes Metab. Res. Rev. 2011, 27, 834–837. [Google Scholar] [CrossRef]
- Beyerlein, A.; Donnachie, E.; Jergens, S.; Ziegler, A.-G. Infections in Early Life and Development of Type 1 Diabetes. JAMA 2016, 315, 1899–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Primavera, M.; Giannini, C.; Chiarelli, F. Prediction and Prevention of Type 1 Diabetes. Front. Endocrinol. 2020, 11, 248. [Google Scholar] [CrossRef]
- Montgomery, S.M.; Ehlin, A.G.C.; Ekbom, A.; Wakefield, A.J. Pertussis infection in childhood and subsequent Type 1 diabetes mellitus. Diabet. Med. 2002, 19, 986–993. [Google Scholar] [CrossRef]
- Bélteky, M.; Wahlberg, J.; Ludvigsson, J. Maternal respiratory infections in early pregnancy increases the risk of type 1 diabetes. Pediatr. Diabetes 2020, 21, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Hippich, M.; Oleynik, A.; Jain, K.; Winkler, C.; Ferreira, R.C.; Bonifacio, E.; Ziegler, A.G.; Briese, T. Searching peripheral blood mononuclear cells of children with viral respiratory tract infections preceding islet autoimmunity for viruses by high-throughput sequencing. Acta Diabetol. 2018, 55, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Beyerlein, A.; Wehweck, F.; Ziegler, A.-G.; Pflueger, M. Respiratory Infections in Early Life and the Development of Islet Autoimmunity in Children at Increased Type 1 Diabetes Risk. JAMA Pediatr. 2013, 167, 800. [Google Scholar] [CrossRef] [Green Version]
- Lynch, K.F.; Lee, H.-S.; Törn, C.; Vehik, K.; Krischer, J.P.; Larsson, H.E.; Haller, M.J.; Hagopian, W.A.; Rewers, M.J.; She, J.-X.; et al. Gestational respiratory infections interacting with offspring HLA and CTLA-4 modifies incident β-cell autoantibodies. J. Autoimmun. 2018, 86, 93–103. [Google Scholar] [CrossRef]
- Harbison, J.E.; Roth-Schulze, A.J.; Giles, L.C.; Tran, C.D.; Ngui, K.M.; Penno, M.A.; Thomson, R.L.; Wentworth, J.M.; Colman, P.G.; Craig, M.E.; et al. Gut microbiome dysbiosis and increased intestinal permeability in children with islet autoimmunity and type 1 diabetes: A prospective cohort study. Pediatr. Diabetes 2019, 20, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Kuitunen, M.; Saukkonen, T.; Ilonen, J.; Åkerblom, H.K.; Savilahti, E. Intestinal Permeability to Mannitol and Lactulose in Children with Type 1 Diabetes with the HLA-DQB1*02 Allele. Autoimmunity 2002, 35, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Secondulfo, M.; Iafusco, D.; Carratù, R.; deMagistris, L.; Sapone, A.; Generoso, M.; Mezzogiomo, A.; Sasso, F.C.; Cartenì, M.; De Rosa, R.; et al. Ultrastructural mucosal alterations and increased intestinal permeability in non-celiac, type I diabetic patients. Dig. Liver Dis. 2004, 36, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Bosi, E.; Molteni, L.; Radaelli, M.G.; Folini, L.; Fermo, I.; Bazzigaluppi, E.; Piemonti, L.; Pastore, M.R.; Paroni, R. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia 2006, 49, 2824–2827. [Google Scholar] [CrossRef] [Green Version]
- Sapone, A.; De Magistris, L.; Pietzak, M.; Clemente, M.G.; Tripathi, A.; Cucca, F.; Lampis, R.; Kryszak, D.; Carteni, M.; Generoso, M.; et al. Zonulin Upregulation Is Associated With Increased Gut Permeability in Subjects With Type 1 Diabetes and Their Relatives. Diabetes 2006, 55, 1443–1449. [Google Scholar] [CrossRef] [Green Version]
- Maffeis, C.; Martina, A.; Corradi, M.; Quarella, S.; Nori, N.; Torriani, S.; Plebani, M.; Contreas, G.; Felis, G.E. Association between intestinal permeability and faecal microbiota composition in Italian children with beta cell autoimmunity at risk for type 1 diabetes. Diabetes Metab. Res. Rev. 2016, 32, 700–709. [Google Scholar] [CrossRef]
- Vaarala, O. The gut as a regulator of early inflammation in type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Vaarala, O. Is the origin of type 1 diabetes in the gut? Immunol. Cell Biol. 2012, 90, 271–276. [Google Scholar] [CrossRef]
- Chonmaitree, T.; Ford, C.; Sanders, C.; Lucia, H.L. Comparison of cell cultures for rapid isolation of enteroviruses. J. Clin. Microbiol. 1988, 26, 2576–2580. [Google Scholar] [CrossRef] [Green Version]
- Shoja, Z.O.; Tabatabie, H.; Shahmahmoudi, S.; Nategh, R. Comparison of cell culture with RT-PCR for enterovirus detection in stool specimens from patients with acute flaccid paralysis. J. Clin. Lab. Anal. 2007, 21, 232–236. [Google Scholar] [CrossRef]
- Isaacs, S.R.; Kim, K.W.; Cheng, J.X.; Bull, R.A.; Stelzer-Braid, S.; Luciani, F.; Rawlinson, W.D.; Craig, M.E. Amplification and next generation sequencing of near full-length human enteroviruses for identification and characterisation from clinical samples. Sci. Rep. 2018, 8, 11889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, R.J.; Wang, J.; Todd, A.K.; Bissielo, A.B.; Yen, S.; Strydom, H.; Moore, N.E.; Ren, X.; Huang, Q.S.; Carter, P.E.; et al. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. J. Virol. Methods 2014, 195, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramna, L.; Kolarova, K.; Oikarinen, S.; Pursiheimo, J.P.; Ilonen, J.; Simell, O.; Knip, M.; Veijola, R.; Hyoty, H.; Cinek, O. Gut virome sequencing in children with early islet autoimmunity. Diabetes Care 2015, 38, 930–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. mBio 2015, 6, e01491-15. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Horton, J.L.; Pang, C.N.I.; Jain, K.; Leung, P.; Isaacs, S.R.; Bull, R.A.; Luciani, F.; Wilkins, M.R.; Catteau, J.; et al. Higher abundance of enterovirus A species in the gut of children with islet autoimmunity. Sci. Rep. 2019, 9, 1749. [Google Scholar] [CrossRef]
- Xu, G.J.; Kula, T.; Xu, Q.; Li, M.Z.; Vernon, S.D.; Ndung’u, T.; Ruxrungtham, K.; Sanchez, J.; Brander, C.; Chung, R.T.; et al. Comprehensive serological profiling of human populations using a synthetic human virome. Science 2015, 348, aaa0698. [Google Scholar] [CrossRef] [Green Version]
- Mina, M.J.; Kula, T.; Leng, Y.; Li, M.; de Vries, R.D.; Knip, M.; Siljander, H.; Rewers, M.; Choy, D.F.; Wilson, M.S.; et al. Measles virus infection diminishes preexisting antibodies that offer protection from other pathogens. Science 2019, 366, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Pou, C.; Nkulikiyimfura, D.; Henckel, E.; Olin, A.; Lakshmikanth, T.; Mikes, J.; Wang, J.; Chen, Y.; Bernhardsson, A.K.; Gustafsson, A.; et al. The repertoire of maternal anti-viral antibodies in human newborns. Nat. Med. 2019, 25, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Schubert, R.D.; Hawes, I.A.; Ramachandran, P.S.; Ramesh, A.; Crawford, E.D.; Pak, J.E.; Wu, W.; Cheung, C.K.; O’Donovan, B.D.; Tato, C.M.; et al. Pan-viral serology implicates enteroviruses in acute flaccid myelitis. Nat. Med. 2019, 25, 1748–1752. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tang, W.; Budhu, A.; Forgues, M.; Hernandez, M.O.; Candia, J.; Kim, Y.; Bowman, E.D.; Ambs, S.; Zhao, Y.; et al. A Viral Exposure Signature Defines Early Onset of Hepatocellular Carcinoma. Cell 2020, 182, 317–328. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [Green Version]
- Handley, S.A. The virome: A missing component of biological interaction networks in health and disease. Genome Med. 2016, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Blanton, L.V.; Cao, S.; Zhao, G.; Manary, M.; Trehan, I.; Smith, M.I.; Wang, D.; Virgin, H.W.; Rohwer, F.; et al. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA 2015, 112, 11941–11946. [Google Scholar] [CrossRef] [Green Version]
- Breitbart, M.; Haynes, M.; Kelley, S.; Angly, F.; Edwards, R.A.; Felts, B.; Mahaffy, J.M.; Mueller, J.; Nulton, J.; Rayhawk, S.; et al. Viral diversity and dynamics in an infant gut. Res. Microbiol. 2008, 159, 367–373. [Google Scholar] [CrossRef]
- Wassenaar, T.M.; Panigrahi, P. Is a foetus developing in a sterile environment? Lett. Appl. Microbiol. 2014, 59, 572–579. [Google Scholar] [CrossRef]
- Liang, G.; Zhao, C.; Zhang, H.; Mattei, L.; Sherrill-Mix, S.; Bittinger, K.; Kessler, L.R.; Wu, G.D.; Baldassano, R.N.; Derusso, P.; et al. The stepwise assembly of the neonatal virome is modulated by breastfeeding. Nature 2020, 581, 470–474. [Google Scholar] [CrossRef]
- Pannaraj, P.S.; Ly, M.; Cerini, C.; Saavedra, M.; Aldrovandi, G.M.; Saboory, A.A.; Johnson, K.M.; Pride, D.T. Shared and Distinct Features of Human Milk and Infant Stool Viromes. Front. Microbiol. 2018, 9, 1162. [Google Scholar] [CrossRef]
- Liang, G.; Bushman, F.D. The human virome: Assembly, composition and host interactions. Nat. Rev. Microbiol. 2021. [Google Scholar] [CrossRef]
- McCann, A.; Ryan, F.J.; Stockdale, S.R.; Dalmasso, M.; Blake, T.; Ryan, C.A.; Stanton, C.; Mills, S.; Ross, P.R.; Hill, C. Viromes of one year old infants reveal the impact of birth mode on microbiome diversity. PeerJ 2018, 6, e4694. [Google Scholar] [CrossRef] [Green Version]
- Maqsood, R.; Rodgers, R.; Rodriguez, C.; Handley, S.A.; Ndao, I.M.; Tarr, P.I.; Warner, B.B.; Lim, E.S.; Holtz, L.R. Discordant transmission of bacteria and viruses from mothers to babies at birth. Microbiome 2019, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Beghetti, I.; Biagi, E.; Martini, S.; Brigidi, P.; Corvaglia, L.; Aceti, A. Human Milk’s Hidden Gift: Implications of the Milk Microbiome for Preterm Infants’ Health. Nutrients 2019, 11, 2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duranti, S.; Lugli, G.A.; Mancabelli, L.; Armanini, F.; Turroni, F.; James, K.; Ferretti, P.; Gorfer, V.; Ferrario, C.; Milani, C.; et al. Maternal inheritance of bifidobacterial communities and bifidophages in infants through vertical transmission. Microbiome 2017, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Makino, H.; Cetinyurek Yavuz, A.; Ben-Amor, K.; Roelofs, M.; Ishikawa, E.; Kubota, H.; Swinkels, S.; Sakai, T.; Oishi, K.; et al. Early-Life Events, Including Mode of Delivery and Type of Feeding, Siblings and Gender, Shape the Developing Gut Microbiota. PLoS ONE 2016, 11, e0158498. [Google Scholar] [CrossRef] [Green Version]
- Holtz, L.R.; Cao, S.; Zhao, G.; Bauer, I.K.; Denno, D.M.; Klein, E.J.; Antonio, M.; Stine, O.C.; Snelling, T.L.; Kirkwood, C.D.; et al. Geographic variation in the eukaryotic virome of human diarrhea. Virology 2014, 468-470, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Song, S.J.; Lauber, C.; Costello, E.K.; Lozupone, C.A.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting family members share microbiota with one another and with their dogs. eLife 2013, 2, e00458. [Google Scholar] [CrossRef]
- Tun, H.M.; Konya, T.; Takaro, T.K.; Brook, J.R.; Chari, R.; Field, C.J.; Guttman, D.S.; Becker, A.B.; Mandhane, P.J.; Turvey, S.E.; et al. Exposure to household furry pets influences the gut microbiota of infants at 3–4 months following various birth scenarios. Microbiome 2017, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penders, J.; Thijs, C.; Vink, C.; Stelma, F.F.; Snijders, B.; Kummeling, I.; Van Den Brandt, P.A.; Stobberingh, E.E. Factors Influencing the Composition of the Intestinal Microbiota in Early Infancy. Pediatrics 2006, 118, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Abeles, S.R.; Ly, M.; Santiago-Rodriguez, T.M.; Pride, D.T. Effects of Long Term Antibiotic Therapy on Human Oral and Fecal Viromes. PLoS ONE 2015, 10, e0134941. [Google Scholar] [CrossRef] [Green Version]
- Kapusinszky, B.; Minor, P.; Delwart, E. Nearly Constant Shedding of Diverse Enteric Viruses by Two Healthy Infants. J. Clin. Microbiol. 2012, 50, 3427–3434. [Google Scholar] [CrossRef] [Green Version]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Wang, D.; Holtz, L.R. The Bacterial Microbiome and Virome Milestones of Infant Development. Trends Microbiol. 2016, 24, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Fulci, V.; Stronati, L.; Cucchiara, S.; Laudadio, I.; Carissimi, C. Emerging Roles of Gut Virome in Pediatric Diseases. Int. J. Mol. Sci. 2021, 22, 4127. [Google Scholar] [CrossRef]
- Lee, H.S.; Briese, T.; Winkler, C.; Rewers, M.; Bonifacio, E.; Hyoty, H.; Pflueger, M.; Simell, O.; She, J.X.; Hagopian, W.; et al. Next-generation sequencing for viruses in children with rapid-onset type 1 diabetes. Diabetologia 2013, 56, 1705–1711. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Vatanen, T.; Droit, L.; Park, A.; Kostic, A.D.; Poon, T.W.; Vlamakis, H.; Siljander, H.; Harkonen, T.; Hamalainen, A.M.; et al. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA 2017, 114, E6166–E6175. [Google Scholar] [CrossRef] [Green Version]
- Cinek, O.; Kramna, L.; Lin, J.; Oikarinen, S.; Kolarova, K.; Ilonen, J.; Simell, O.; Veijola, R.; Autio, R.; Hyoty, H. Imbalance of bacteriome profiles within the Finnish Diabetes Prediction and Prevention study: Parallel use of 16S profiling and virome sequencing in stool samples from children with islet autoimmunity and matched controls. Pediatr. Diabetes 2017, 18, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Allen, D.W.; Briese, T.; Couper, J.J.; Barry, S.C.; Colman, P.G.; Cotterill, A.M.; Davis, E.A.; Giles, L.C.; Harrison, L.C.; et al. Distinct Gut Virome Profile of Pregnant Women With Type 1 Diabetes in the ENDIA Study. Open Forum Infect. Dis. 2019, 6, ofz025. [Google Scholar] [CrossRef]
- Kim, K.W.; Allen, D.W.; Briese, T.; Couper, J.J.; Barry, S.C.; Colman, P.G.; Cotterill, A.M.; Davis, E.A.; Giles, L.C.; Harrison, L.C.; et al. Higher frequency of vertebrate-infecting viruses in the gut of infants born to mothers with type 1 diabetes. Pediatr. Diabetes 2020, 21, 271–279. [Google Scholar] [CrossRef]
- Cinek, O.; Kramna, L.; Odeh, R.; Alassaf, A.; Ibekwe, M.A.U.; Ahmadov, G.; Elmahi, B.M.E.; Mekki, H.; Lebl, J.; Abdullah, M.A. Eukaryotic viruses in the fecal virome at the onset of type 1 diabetes: A study from four geographically distant African and Asian countries. Pediatr. Diabetes 2021, 22, 558–566. [Google Scholar] [CrossRef]
- Kim, K.W.; Deveson, I.W.; Pang, C.N.I.; Yeang, M.; Naing, Z.; Adikari, T.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Verich, A.; et al. Respiratory viral co-infections among SARS-CoV-2 cases confirmed by virome capture sequencing. Sci. Rep. 2021, 11, 3934. [Google Scholar] [CrossRef]
- Sutton, T.D.S.; Clooney, A.G.; Ryan, F.J.; Ross, R.P.; Hill, C. Choice of assembly software has a critical impact on virome characterisation. Microbiome 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Calvo, T.; von Herrath, M.G. Enterovirus Infection and Type 1 Diabetes: Closing in on a Link? Diabetes 2015, 64, 1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunne, J.L.; Richardson, S.J.; Atkinson, M.A.; Craig, M.E.; Dahl-Jorgensen, K.; Flodstrom-Tullberg, M.; Hyoty, H.; Insel, R.A.; Lernmark, A.; Lloyd, R.E.; et al. Rationale for enteroviral vaccination and antiviral therapies in human type 1 diabetes. Diabetologia 2019, 62, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, V.M.; Hankaniemi, M.M.; Svedin, E.; Sioofy-Khojine, A.; Oikarinen, S.; Hyoty, H.; Laitinen, O.H.; Hytonen, V.P.; Flodstrom-Tullberg, M. A Coxsackievirus B vaccine protects against virus-induced diabetes in an experimental mouse model of type 1 diabetes. Diabetologia 2018, 61, 476–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, V.M.; Hankaniemi, M.M.; Laitinen, O.H.; Sioofy-Khojine, A.B.; Lin, A.; Diaz Lozano, I.M.; Mazur, M.A.; Marjomäki, V.; Loré, K.; Hyöty, H.; et al. A hexavalent Coxsackievirus B vaccine is highly immunogenic and has a strong protective capacity in mice and nonhuman primates. Sci. Adv. 2020, 6, eaaz2433. [Google Scholar] [CrossRef]
- Chumakov, K.; Ehrenfeld, E.; Plotkin, S. New Generation of Inactivated Poliovirus Vaccines for Universal Immunization after Eradication of Poliomyelitis. Clin. Infect. Dis. 2008, 47, 1587–1592. [Google Scholar] [CrossRef] [Green Version]
- Hankaniemi, M.M.; Stone, V.M.; Andrejeff, T.; Heinimäki, S.; Sioofy-Khojine, A.-B.; Marjomäki, V.; Hyöty, H.; Blazevic, V.; Flodström-Tullberg, M.; Hytönen, V.P.; et al. Formalin treatment increases the stability and immunogenicity of coxsackievirus B1 VLP vaccine. Antivir. Res. 2019, 171, 104595. [Google Scholar] [CrossRef]
- Koho, T.; Koivunen, M.R.L.; Oikarinen, S.; Kummola, L.; Mäkinen, S.; Mähönen, A.J.; Sioofy-Khojine, A.; Marjomäki, V.; Kazmertsuk, A.; Junttila, I.; et al. Coxsackievirus B3 VLPs purified by ion exchange chromatography elicit strong immune responses in mice. Antivir. Res. 2014, 104, 93–101. [Google Scholar] [CrossRef]
- Hankaniemi, M.M.; Baikoghli, M.A.; Stone, V.M.; Xing, L.; Väätäinen, O.; Soppela, S.; Sioofy-Khojine, A.; Saarinen, N.V.V.; Ou, T.; Anson, B.; et al. Structural Insight into CVB3-VLP Non-Adjuvanted Vaccine. Microorganisms 2020, 8, 1287. [Google Scholar] [CrossRef]
- Hassine, I.H.; Gharbi, J.; Hamrita, B.; Almalki, M.A.; Rodríguez, J.F.; Ben M’hadheb, M. Characterization of Coxsackievirus B4 virus-like particles VLP produced by the recombinant baculovirus-insect cell system expressing the major capsid protein. Mol. Biol. Rep. 2020, 47, 2835–2843. [Google Scholar] [CrossRef]
- Anasir, M.I.; Zarif, F.; Poh, C.L. Antivirals blocking entry of enteroviruses and therapeutic potential. J. Biomed. Sci. 2021, 28, 10. [Google Scholar] [CrossRef] [PubMed]
- Pevear, D.C.; Tull, T.M.; Seipel, M.E.; Groarke, J.M. Activity of pleconaril against enteroviruses. Antimicrob. Agents Chemother. 1999, 43, 2109–2115. [Google Scholar] [CrossRef] [Green Version]
- Honkimaa, A.; Sioofy-Khojine, A.B.; Oikarinen, S.; Bertin, A.; Hober, D.; Hyöty, H. Eradication of persistent coxsackievirus B infection from a pancreatic cell line with clinically used antiviral drugs. J. Clin. Virol. 2020, 128, 104334. [Google Scholar] [CrossRef] [PubMed]
- Alidjinou, E.K.; Sané, F.; Bertin, A.; Caloone, D.; Hober, D. Persistent infection of human pancreatic cells with Coxsackievirus B4 is cured by fluoxetine. Antivir. Res. 2015, 116, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, C.; Kim, D.E.; Song, J.H.; Choi, M.; Choi, K.; Kang, M.; Lee, K.; Kim, H.S.; Shin, J.S.; et al. Synergistic antiviral activity of gemcitabine and ribavirin against enteroviruses. Antivir. Res. 2015, 124, 1–10. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isaacs, S.R.; Foskett, D.B.; Maxwell, A.J.; Ward, E.J.; Faulkner, C.L.; Luo, J.Y.X.; Rawlinson, W.D.; Craig, M.E.; Kim, K.W. Viruses and Type 1 Diabetes: From Enteroviruses to the Virome. Microorganisms 2021, 9, 1519. https://doi.org/10.3390/microorganisms9071519
Isaacs SR, Foskett DB, Maxwell AJ, Ward EJ, Faulkner CL, Luo JYX, Rawlinson WD, Craig ME, Kim KW. Viruses and Type 1 Diabetes: From Enteroviruses to the Virome. Microorganisms. 2021; 9(7):1519. https://doi.org/10.3390/microorganisms9071519
Chicago/Turabian StyleIsaacs, Sonia R., Dylan B. Foskett, Anna J. Maxwell, Emily J. Ward, Clare L. Faulkner, Jessica Y. X. Luo, William D. Rawlinson, Maria E. Craig, and Ki Wook Kim. 2021. "Viruses and Type 1 Diabetes: From Enteroviruses to the Virome" Microorganisms 9, no. 7: 1519. https://doi.org/10.3390/microorganisms9071519
APA StyleIsaacs, S. R., Foskett, D. B., Maxwell, A. J., Ward, E. J., Faulkner, C. L., Luo, J. Y. X., Rawlinson, W. D., Craig, M. E., & Kim, K. W. (2021). Viruses and Type 1 Diabetes: From Enteroviruses to the Virome. Microorganisms, 9(7), 1519. https://doi.org/10.3390/microorganisms9071519