
Microbiomes of Various Maternal Body Systems Are Predictive of Calf Digestive Bacterial Ecology

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Observation and Sample Collection
2.2. Serum and Colostrum IgG
2.3. DNA Extraction and Sequencing
2.4. Bioinformatics Analysis
2.4.1. Taxonomic Profiling
2.4.2. Alpha and Beta Diversity
2.4.3. Microbiome Associations
3. Results
3.1. Descriptive Statistics
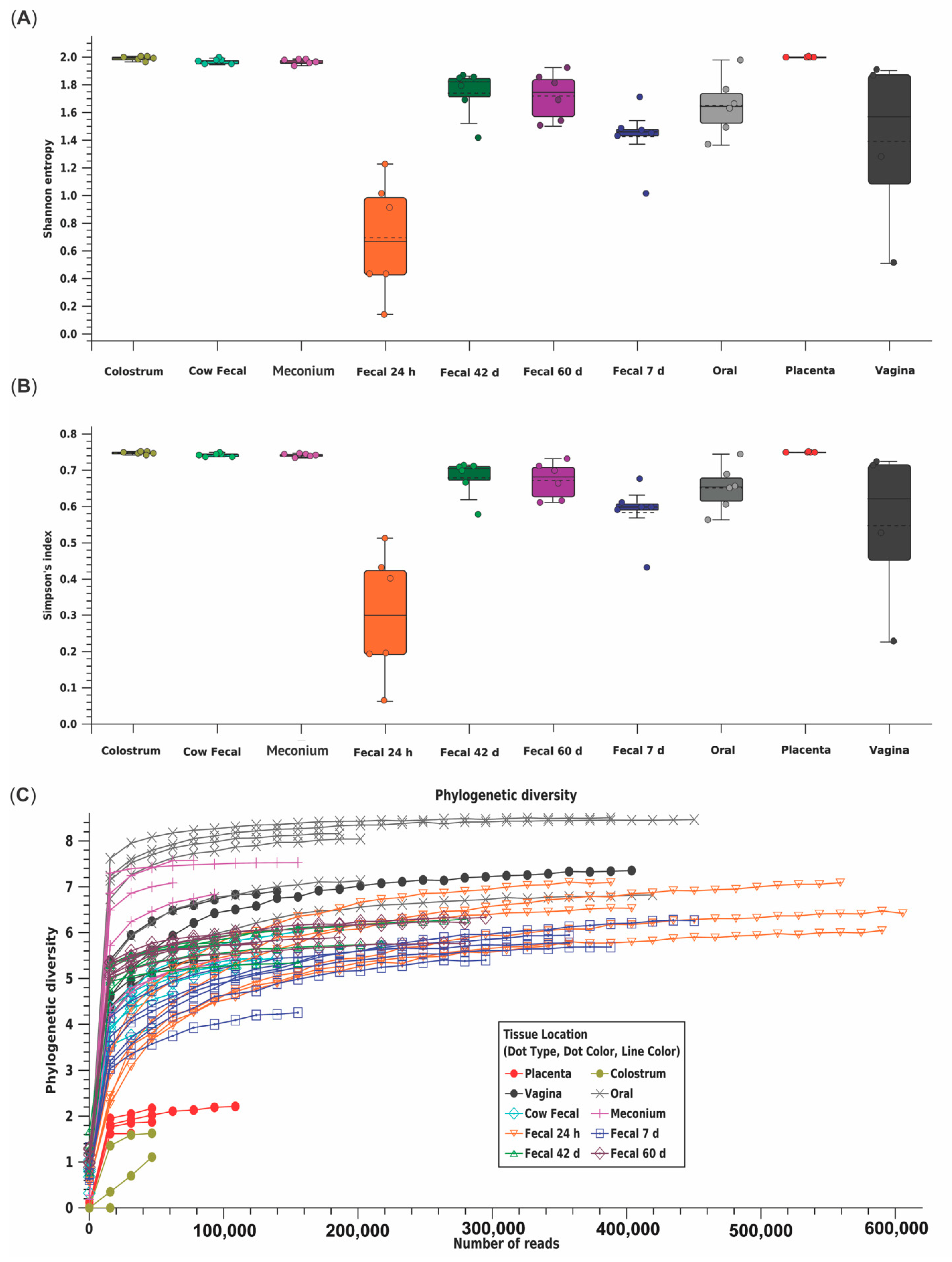
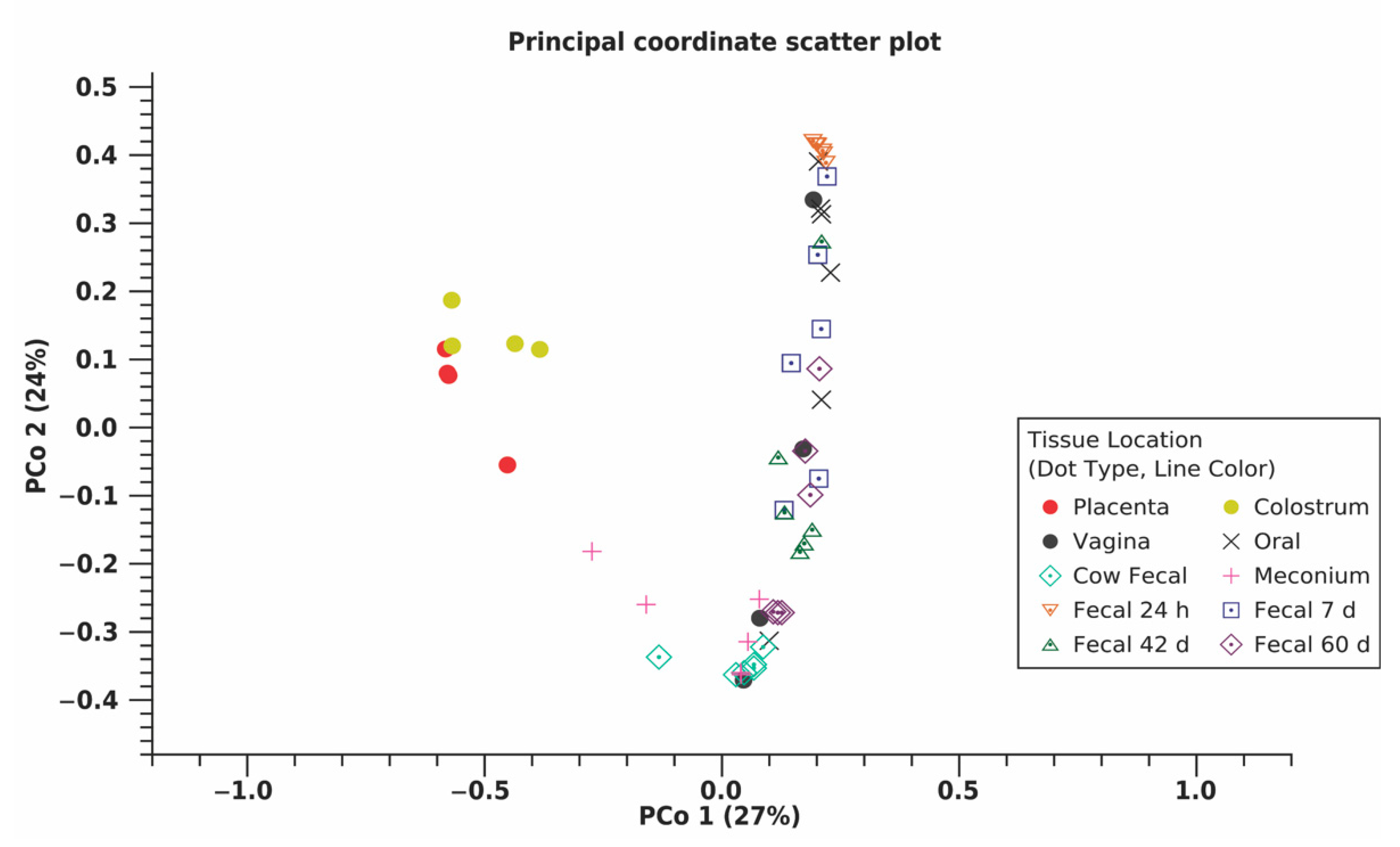
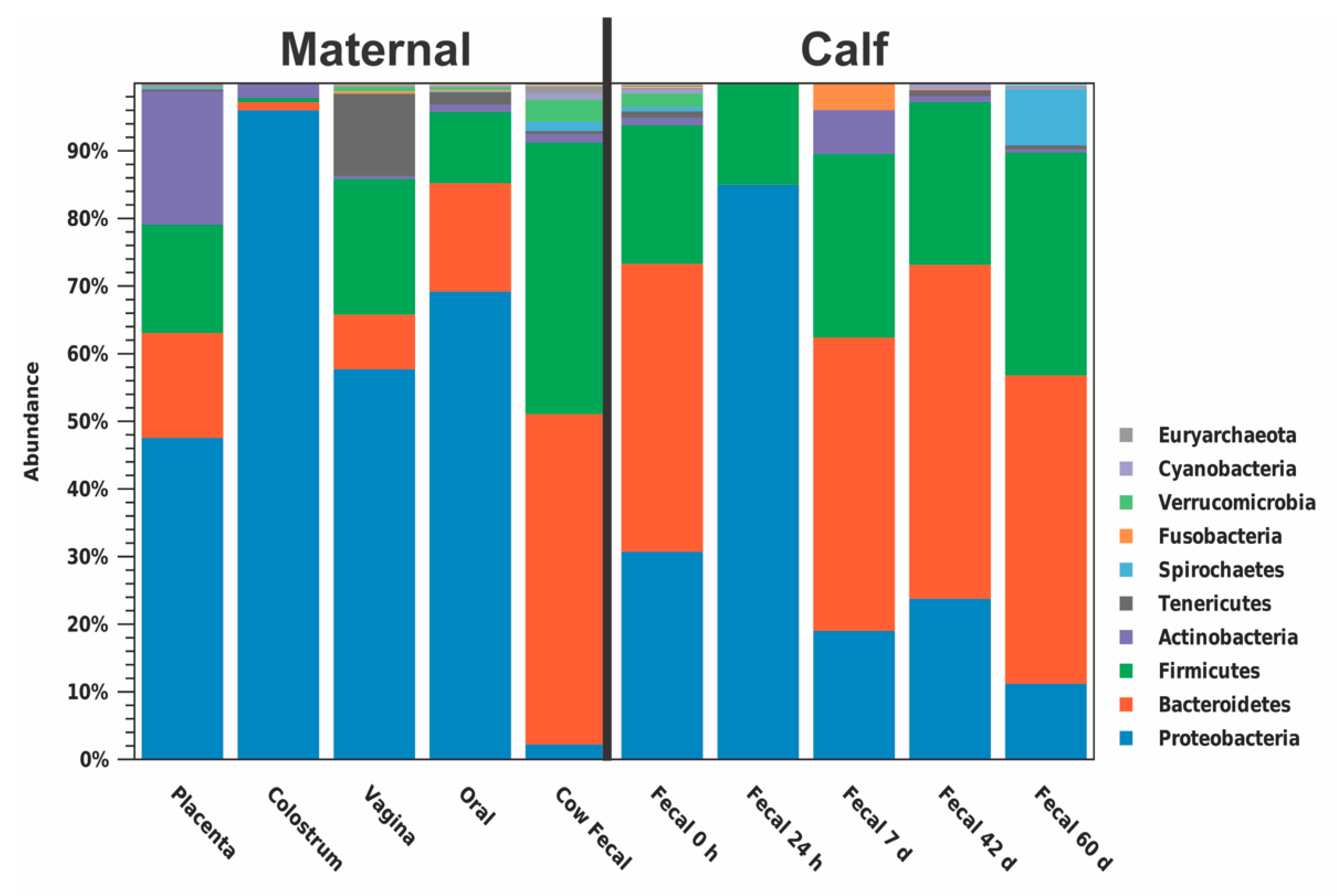
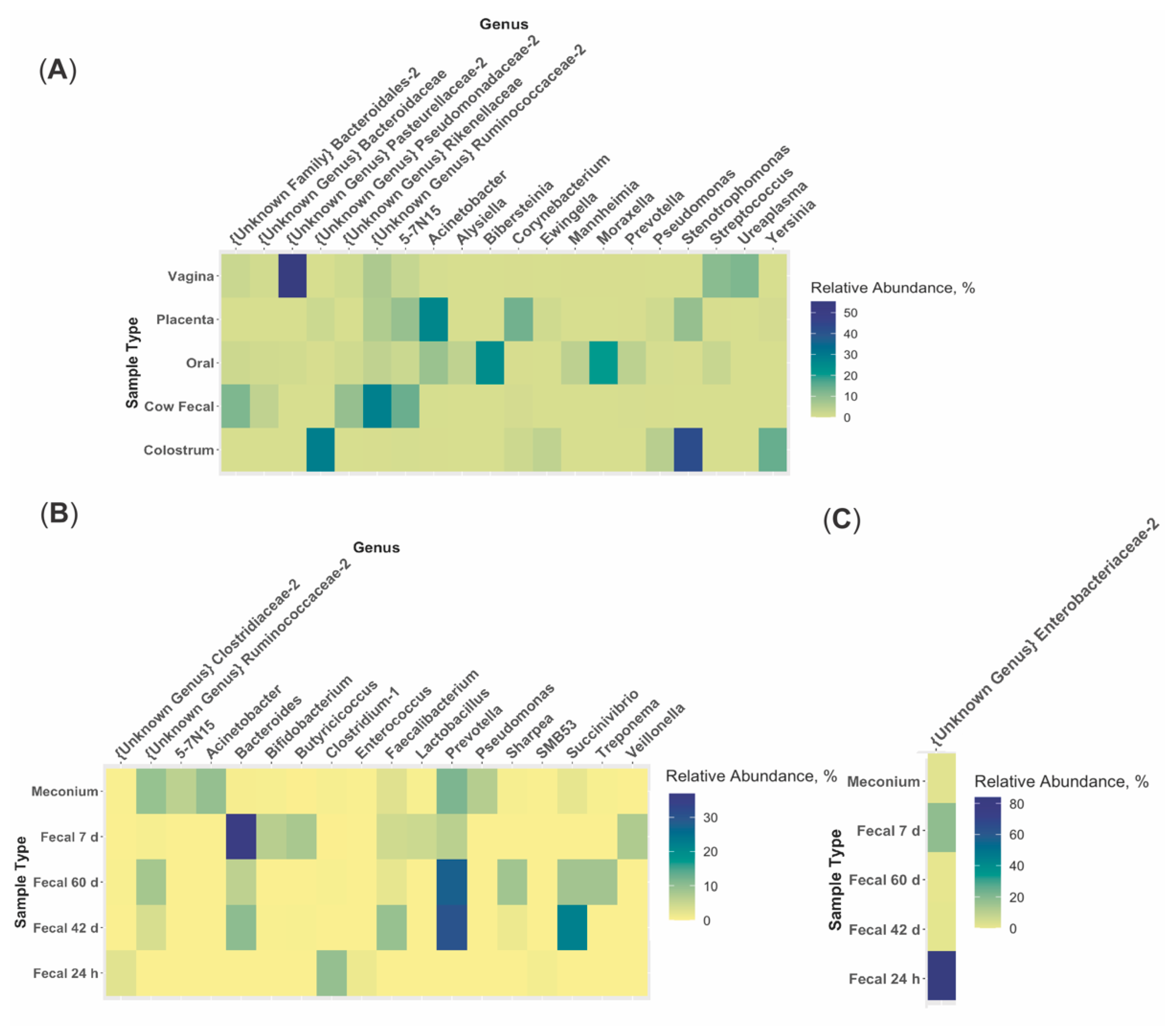
3.2. Bioinformatics Analyses
3.3. Microbiome Associations
4. Discussion
4.1. Early Changes in the Calf Fecal Microbiome
4.2. Variation between Maternal Sources
4.3. Microbiome Heritability
4.4. Study Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Malmuthuge, N.; Li, M.; Fries, P.; Griebel, P.J.; Guan, L.L. Regional and age dependent changes in gene expression of toll-like receptors and key antimicrobial defense molecules throughout the gastrointestinal tract of dairy calves. Vet. Immunol. Immunopathol. 2012, 146, 18–26. [Google Scholar] [CrossRef]
- Maynard, C.L.; Elson, C.O.; Hatton, R.D.; Weaver, C.T. Reciprocal interactions of the intestinal microbiota and immune system. Nature 2012, 489, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.; Abenthum, A.; Matthes, J.M.; Kleeberger, D.; Ege, M.J.; Hölzel, C.; Bauer, J.; Schwaiger, K. Development and genetic influence of the rectal bacterial flora of newborn calves. Vet. Microbiol. 2012, 161, 179–185. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Peng, D.; Li, G.; Chen, J.; Gu, X. Exposure to heat-stress environment affects the physiology, circulation levels of cytokines, and microbiome in dairy cows. Sci. Rep. 2018, 8, 14606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyte, M. The effect of stress on microbial growth. Anim. Health Res. Rev. 2014, 15, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.E.; Arroyo, L.G.; Costa, M.C.; Viel, L.; Weese, J.S. Characterization of the fecal bacterial microbiota of healthy and diarrheic dairy calves. J. Vet. Intern. Med. 2017, 31, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, J.M.; Kieran, T.J.; Seidel, D.S.; Glenn, T.C.; Silveira, M.F.d.; Callaway, T.R.; Stewart, R.L., Jr. Comparison of the ruminal and fecal microbiotas in beef calves supplemented or not with concentrate. PLoS ONE 2020, 15, e0231533. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Villot, C.; Renaud, D.; Skidmore, A.; Chevaux, E.; Steele, M. Linking perturbations to temporal changes in diversity, stability, and compositions of neonatal calf gut microbiota: Prediction of diarrhea. ISME J. 2020, 14, 1–13. [Google Scholar] [CrossRef]
- Klein-Jöbstl, D.; Quijada, N.M.; Dzieciol, M.; Feldbacher, B.; Wagner, M.; Drillich, M.; Schmitz-Esser, S.; Mann, E. Microbiota of newborn calves and their mothers reveals possible transfer routes for newborn calves’ gastrointestinal microbiota. PLoS ONE 2019, 14, e0220554. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, H.C.; Austin, K.J.; Powell, S.R.; Carpenter, K.T.; Cammack, K.M. Potential response of the rumen microbiome to mode of delivery from birth through weaning. Transl. Anim. Sci. 2018, 2, S35–S38. [Google Scholar] [CrossRef] [PubMed]
- Yeoman, C.J.; Ishaq, S.L.; Bichi, E.; Olivo, S.K.; Lowe, J.; Aldridge, B.M. Biogeographical differences in the influence of maternal microbial sources on the early successional development of the bovine neonatal gastrointestinal tract. Sci. Rep. 2018, 8, 3197. [Google Scholar] [CrossRef]
- Rodríguez, J.M. The origin of human milk bacteria: Is there a bacterial entero-mammary pathway during late pregnancy and lactation? Adv. Nutr. 2014, 5, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Guzman, C.E.; Wood, J.L.; Egidi, E.; White-Monsant, A.C.; Semenec, L.; Grommen, S.V.H.; Hill-Yardin, E.L.; de Groef, B.; Franks, A.E. A pioneer calf foetus microbiome. Sci. Rep. 2020, 10, 17712. [Google Scholar] [CrossRef]
- Moore, S.G.; Ericsson, A.C.; Poock, S.E.; Melendez, P.; Lucy, M.C. Hot topic: 16S rRNA gene sequencing reveals the microbiome of the virgin and pregnant bovine uterus. J. Dairy Sci. 2017, 100, 4953–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, M.; Veerus, L.; Trosvik, P.; Buckling, A.; Pizzari, T. The reproductive microbiome: An emerging driver of sexual selection, sexual conflict, mating systems, and reproductive isolation. Trends Ecol. Evol. 2020, 35, 220–234. [Google Scholar] [CrossRef] [Green Version]
- Young, W.; Hine, B.C.; Wallace, O.A.; Callaghan, M.; Bibiloni, R. Transfer of intestinal bacterial components to mammary secretions in the cow. PeerJ 2015, 3, e888. [Google Scholar] [CrossRef] [PubMed]
- Erbilgin, O.; Bowen, B.P.; Kosina, S.M.; Jenkins, S.; Lau, R.K.; Northen, T.R. Dynamic substrate preferences predict metabolic properties of a simple microbial consortium. BMC Bioinform. 2017, 18, 57. [Google Scholar] [CrossRef] [Green Version]
- Tuncil, Y.E.; Xiao, Y.; Porter, N.T.; Reuhs, B.L.; Martens, E.C.; Hamaker, B.R. Reciprocal prioritization to dietary glycans by gut bacteria in a competitive environment promotes stable coexistence. mBio 2017, 8, e01068-17. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the illumina hiseq and miseq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Estimating the pattern of nucleotide substitution. J. Mol. Evol. 1994, 39, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.R-project.org/ (accessed on 15 January 2020).
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002; ISBN 0-387-95457-0. Available online: http://www.stats.ox.ac.uk/pub/MASS4 (accessed on 15 January 2020).
- Alipour, M.J.; Jalanka, J.; Pessa-Morikawa, T.; Kokkonen, T.; Satokari, R.; Hynönen, U.; Iivanainen, A.; Niku, M. The composition of the perinatal intestinal microbiota in cattle. Sci. Rep. 2018, 8, 10437. [Google Scholar] [CrossRef]
- Rey, M.; Enjalbert, F.; Combes, S.; Cauquil, L.; Bouchez, O.; Monteils, V. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J. Appl. Microbiol. 2014, 116, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Wilson, M. Microbial Inhabitants of Humans: Their Ecology and Role in Health and Disease; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Chow, W.L.; Lee, Y.-K. Mucosal interactions and gastrointestinal microbiota. In Gastrointestinal Microbiology, 1st ed.; Ouwehand, A.C., Vaughan, E.E., Eds.; CRC Press: New York City, NY, USA, 2006; Volume 1, pp. 123–136. [Google Scholar]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111. [Google Scholar] [CrossRef] [Green Version]
- Redford, A.J.; Fierer, N. Bacterial succession on the leaf Surface: A novel system for studying successional dynamics. Microb. Ecol. 2009, 58, 189–198. [Google Scholar] [CrossRef]
- Pascault, N.; Roux, S.; Artigas, J.; Pesce, S.; Leloup, J.; Tadonleke, R.D.; Debroas, D.; Bouchez, A.; Humbert, J.-F. A high-throughput sequencing ecotoxicology study of freshwater bacterial communities and their responses to tebuconazole. FEMS Microbiol. Ecol. 2014, 90, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.; Mann, A.J.; Waldmann, J.; et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 2012, 336, 608. [Google Scholar] [CrossRef]
- Whon, T.W.; Kim, M.-S.; Roh, S.W.; Shin, N.-R.; Lee, H.-W.; Bae, J.-W. Metagenomic characterization of airborne viral DNA diversity in the near-surface atmosphere. J. Virol. 2012, 86, 8221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iljazovic, A.; Roy, U.; Gálvez, E.J.C.; Lesker, T.R.; Zhao, B.; Gronow, A.; Amend, L.; Will, S.E.; Hofmann, J.D.; Pils, M.C.; et al. Perturbation of the gut microbiome by prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal Immunol. 2021, 14, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Whon Tae, W.; Hyun, S.K.; Shin, N.-R.; Sung, H.; Kim, M.-S.; Joon, Y.K.; Kang, W.; Pil, S.K.; Hyun, D.-W.; Hoon, J.S.; et al. Calf diarrhea caused by prolonged expansion of autochthonous gut enterobacteriaceae and their lytic bacteriophages. mSystems 2021, 6, e00816-20. [Google Scholar] [CrossRef]
- Jiménez, E.; Fernández, L.; Marín, M.L.; Martín, R.; Odriozola, J.M.; Nueno-Palop, C.; Narbad, A.; Olivares, M.; Xaus, J.; Rodríguez, J.M. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr. Microbiol. 2005, 51, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Kuroda, Y.; Sugiyama, A. A comparison of the histological structure of the placenta in experimental animals. J. Toxicol. Pathol. 2014, 27, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, E.D.; Cross, J.C. Development of structures and transport functions in the mouse placenta. Physiology 2005, 20, 180–193. [Google Scholar] [CrossRef]
- Dunlap, K.A.; Brown, J.D.; Keith, A.B.; Satterfield, M.C. Factors controlling nutrient availability to the developing fetus in ruminants. J. Anim. Sci. Biotechnol. 2015, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Barden, M.; Richards-Rios, P.; Ganda, E.; Lenzi, L.; Eccles, R.; Neary, J.; Oultram, J.; Oikonomou, G. Maternal influences on oral and faecal microbiota maturation in neonatal calves in beef and dairy production systems. Anim. Microbiome 2020, 2, 31. [Google Scholar] [CrossRef]
- Wang, Y.; Ametaj, B.N.; Ambrose, D.J.; Gänzle, M.G. Characterisation of the bacterial microbiota of the vagina of dairy cows and isolation of pediocin-producing pediococcus acidilactici. BMC Microbiol. 2013, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Appiah, M.O.; Wang, J.; Lu, W. Microflora in the reproductive tract of cattle: A review. Agriculture 2020, 10, 232. [Google Scholar] [CrossRef]
- Owens, C.E.; Daniels, K.M.; Ealy, A.D.; Knowlton, K.F.; Cockrum, R.R. Graduate student literature review: Potential mechanisms of interaction between bacteria and the reproductive tract of dairy cattle. J. Dairy Sci. 2020, 103, 10951–10960. [Google Scholar] [CrossRef]
- Russell, J.B.; Baldwin, R.L. Substrate preferences in rumen bacteria: Evidence of catabolite regulatory mechanisms. Appl. Environ. Microbiol. 1978, 36, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasson, G.; Ben-Shabat, S.K.; Seroussi, E.; Doron-Faigenboim, A.; Shterzer, N.; Yaacoby, S.; Miller, M.E.B.; White, B.A.; Halperin, E.; Mizrahi, I. Heritable bovine rumen bacteria are phylogenetically related and correlated with the cow’s capacity to harvest energy from its feed. mBio 2017, 8, e00703–e00717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, C.; Chen, Y.; Liu, J.; Zhang, C.; Irving, B.; Fitzsimmons, C.; Plastow, G.; Guan, L.L. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 2019, 7, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef] [Green Version]
- Senoo, H.; Hata, R. Extracellular matrix regulates cell morphology, proliferation, and tissue formation. Kaibogaku zasshi. J. Anat. 1994, 69, 719–733. [Google Scholar]
- Ash, J.T.; Darnell, G.; Munro, D.; Engelhardt, B.E. Joint analysis of gene expression levels and histological images identifies genes associated with tissue morphology. Nat. Commun. 2021, 12, 1609. [Google Scholar] [CrossRef]
- Mulder, I.E.; Schmidt, B.; Stokes, C.R.; Lewis, M.; Bailey, M.; Aminov, R.I.; Prosser, J.I.; Gill, B.P.; Pluske, J.R.; Mayer, C.D.; et al. Environmentally-acquired bacteria influence microbial diversity and natural innate immune responses at gut surfaces. BMC Biol. 2009, 7, 79. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Mean | SEM | Min | Max |
|---|---|---|---|---|
| Birth weight, kg | 46.00 | 0.94 | 43.00 | 48.00 |
| ADG, kg/d | 0.70 | 0.03 | 0.61 | 0.76 |
| Water intake, kg/d | 0.60 | 0.06 | 0.41 | 0.82 |
| Feed intake, kg/d | 0.61 | 0.02 | 0.54 | 0.68 |
| Fecal score, 0–3 2 | 0.50 | 0.00 | 0.25 | 0.75 |
| Colostrum brix, % 3 | 26 | 0.88 | 23 | 29 |
| Colostrum volume, L | 6.5 | 0.86 | 3.8 | 9.5 |
| Colostrum IgG, mg/dL | 13,502 | 1976 | 5696 | 18,570 |
| Calf serum IgG, mg/dL 4 | 2997 | 251 | 1783 | 4388 |
| Tissue | Total Reads | Reads in OTU 1 | Number of OTU 1 |
|---|---|---|---|
| Placenta | 385,350 ± 26,534 | 61,688 ± 15,016 | 187.00 ± 56.44 |
| Colostrum | 362,749 ± 49,633 | 32,776 ± 6923 | 20.50 ± 7.42 |
| Vagina | 314,095 ± 57,108 | 184,739 ± 74,332 | 1436.25 ± 114.11 |
| Oral | 431,700 ± 58,540 | 313,742 ± 50,289 | 2202.67 ± 466.09 |
| Dam Fecal | 204,011 ± 29,443 | 110,857 ± 16,556 | 1498.83 ± 176.39 |
| Meconium | 244,533 ± 16,662 | 107,453 ± 14,227 | 1223.33 ± 146.94 |
| 24 h Fecal | 543,118 ± 51,403 | 490,960 ± 44,588 | 339.33 ± 29.01 |
| 7 d Fecal | 372,971 ± 44,982 | 338,514 ± 41,614 | 406.00 ± 35.87 |
| 42 d Fecal | 208,827 ± 29,414 | 176,874 ± 27,221 | 797.33 ± 27.06 |
| 60 d Fecal | 262,002 ± 40,738 | 216,843 ± 33,375 | 1063.83 ± 95.34 |
| Placenta | Vagina 2 | Oral | Fecal | Meconium | 24 h | 7 d | 42 d | 60 d | |
|---|---|---|---|---|---|---|---|---|---|
| Colostrum | 0.175 | 0.121 | 0.110 | 0.056 | 0.128 | 0.097 | 0.073 | 0.052 | 0.050 |
| Placenta | 0.306 | 0.292 | 0.228 | 0.267 | 0.221 | 0.204 | 0.193 | 0.210 | |
| Vagina 2 | 0.463 | 0.506 | 0.142 | 0.312 | 0.309 | 0.329 | 0.404 | ||
| Oral | 0.432 | 0.527 | 0.310 | 0.309 | 0.319 | 0.347 | |||
| Fecal | 0.337 | 0.329 | 0.335 | 0.420 | 0.477 | ||||
| Meconium | 0.276 | 0.282 | 0.347 | 0.360 | |||||
| 24 h | 0.632 | 0.402 | 0.410 | ||||||
| 7 d | 0.523 | 0.473 | |||||||
| 42 d | 0.729 |
| Maternal Location | Meconium | 24 h Fecal | 7 d Fecal | 42 d Fecal | 60 d Fecal | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Estimate 3 | p | Estimate | p | Estimate | p | Estimate | p | Estimate | p | |
| Placenta | 4.96 × 10−4 | 0.390 | 1.87 × 10−2 | <0.001 | −3.02 × 10−3 | <0.001 | −1.75 × 10−3 | 0.27 | 1.72 × 10−4 | 0.722 |
| Colostrum | 2.72 × 10−3 | <0.001 | −4.26 × 10−3 | <0.001 | 8.58 × 10−2 | 0.001 | −1.81 × 10−2 | 0.180 | −2.30 × 10−3 | 0.003 |
| Vagina 2 | 1.23 × 10−5 | 0.130 | −2.29 × 10−6 | 0.770 | −1.79 × 10−5 | 0.025 | −2.93 × 10−5 | 0.180 | −1.70 × 10−5 | 0.014 |
| Oral | 1.73 × 10−4 | <0.001 | −7.70 × 10−6 | 0.100 | 3.31 × 10−5 | <0.001 | 5.57 × 10−4 | <0.001 | 2.78 × 10−5 | <0.001 |
| Fecal | 1.59 × 10−4 | <0.001 | −5.90 × 10−5 | <0.001 | −2.34 × 10−6 | 0.870 | 4.78 × 10−5 | 0.201 | 1.19 × 10−5 | 0.312 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Owens, C.E.; Huffard, H.G.; Nin-Velez, A.I.; Duncan, J.; Teets, C.L.; Daniels, K.M.; Ealy, A.D.; James, R.E.; Knowlton, K.F.; Cockrum, R.R. Microbiomes of Various Maternal Body Systems Are Predictive of Calf Digestive Bacterial Ecology. Animals 2021, 11, 2210. https://doi.org/10.3390/ani11082210
Owens CE, Huffard HG, Nin-Velez AI, Duncan J, Teets CL, Daniels KM, Ealy AD, James RE, Knowlton KF, Cockrum RR. Microbiomes of Various Maternal Body Systems Are Predictive of Calf Digestive Bacterial Ecology. Animals. 2021; 11(8):2210. https://doi.org/10.3390/ani11082210
Chicago/Turabian StyleOwens, Connor E., Haley G. Huffard, Alexandra I. Nin-Velez, Jane Duncan, Chrissy L. Teets, Kristy M. Daniels, Alan D. Ealy, Robert E. James, Katharine F. Knowlton, and Rebecca R. Cockrum. 2021. "Microbiomes of Various Maternal Body Systems Are Predictive of Calf Digestive Bacterial Ecology" Animals 11, no. 8: 2210. https://doi.org/10.3390/ani11082210