
Identification of Novel lncRNA and Differentially Expressed Genes (DEGs) of Testicular Tissues among Cattle, Yak, and Cattle-Yak Associated with Male Infertility


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statements
2.2. Sample Collection and Preparation
2.3. RNA Extraction, Library Preparation, and Illumina Sequencing
2.4. Quality Control, Mapping, and Transcriptome Assembly
2.5. Quantification of Gene Expression Levels
2.6. Unknown Transcripts Prediction and LncRNA Analysis
2.7. Target Genes Prediction and Differential Expression Analysis
2.8. Functional Enrichment Analysis
2.9. qPCR Validation and Statistical Analysis
3. Result
3.1. Sequencing and Reads Mapping
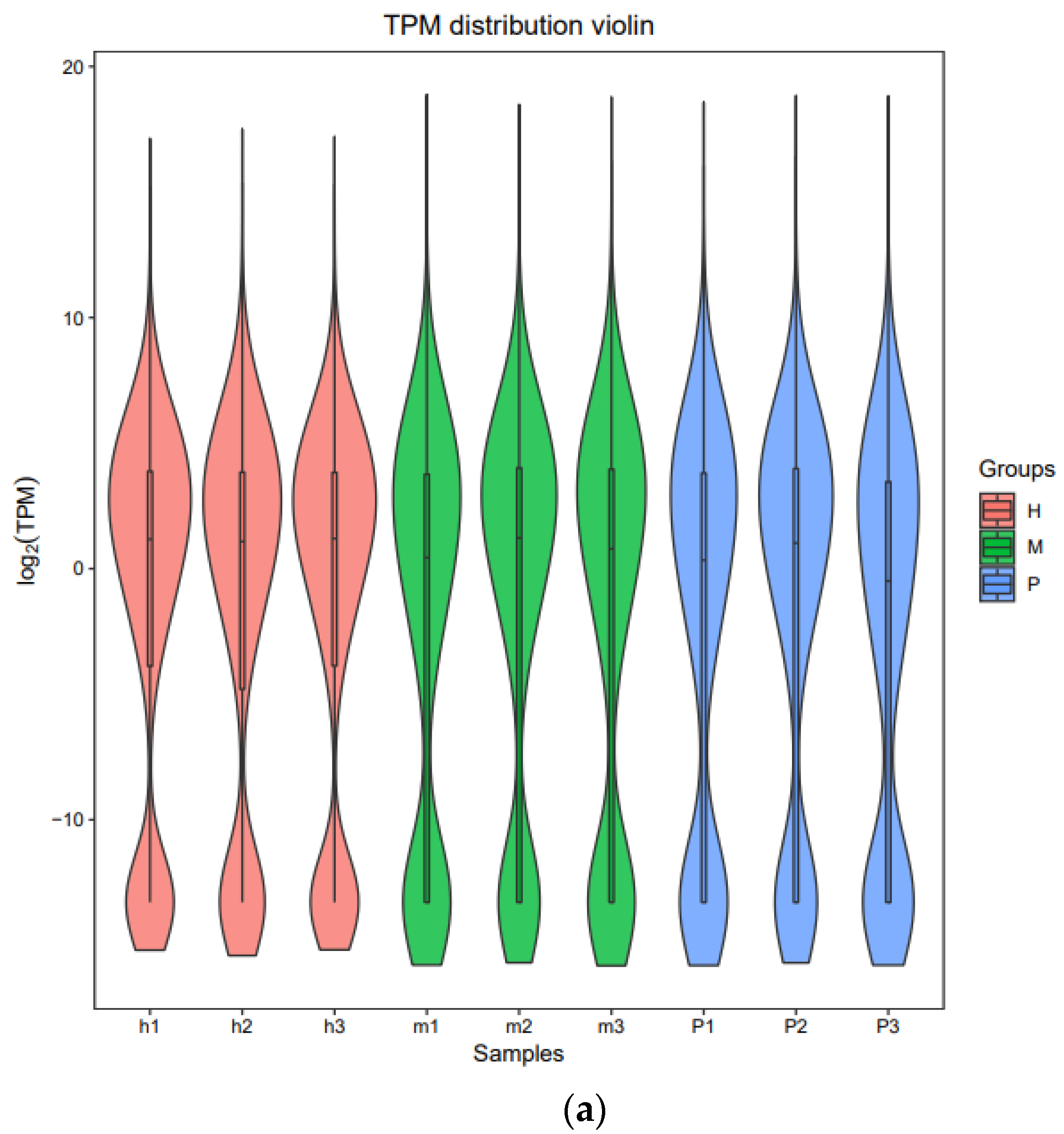
3.2. Quantification of Gene Expression Levels
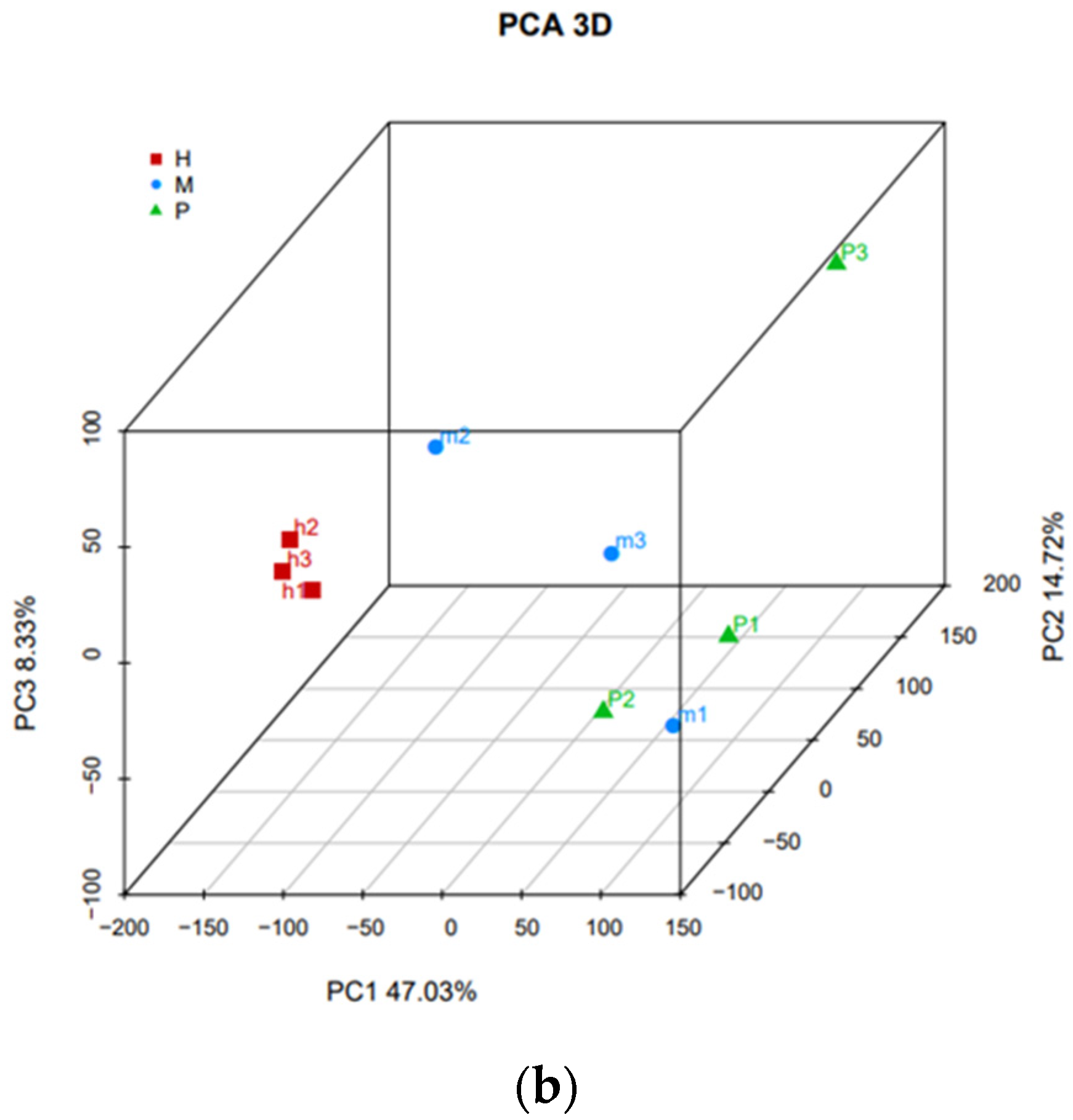
3.3. Differential Analysis between Three Groups and Nine Samples
3.4. LncRNA Screening
3.5. Prediction of lncRNA Target Genes and Differential Expression (DE) Analysis
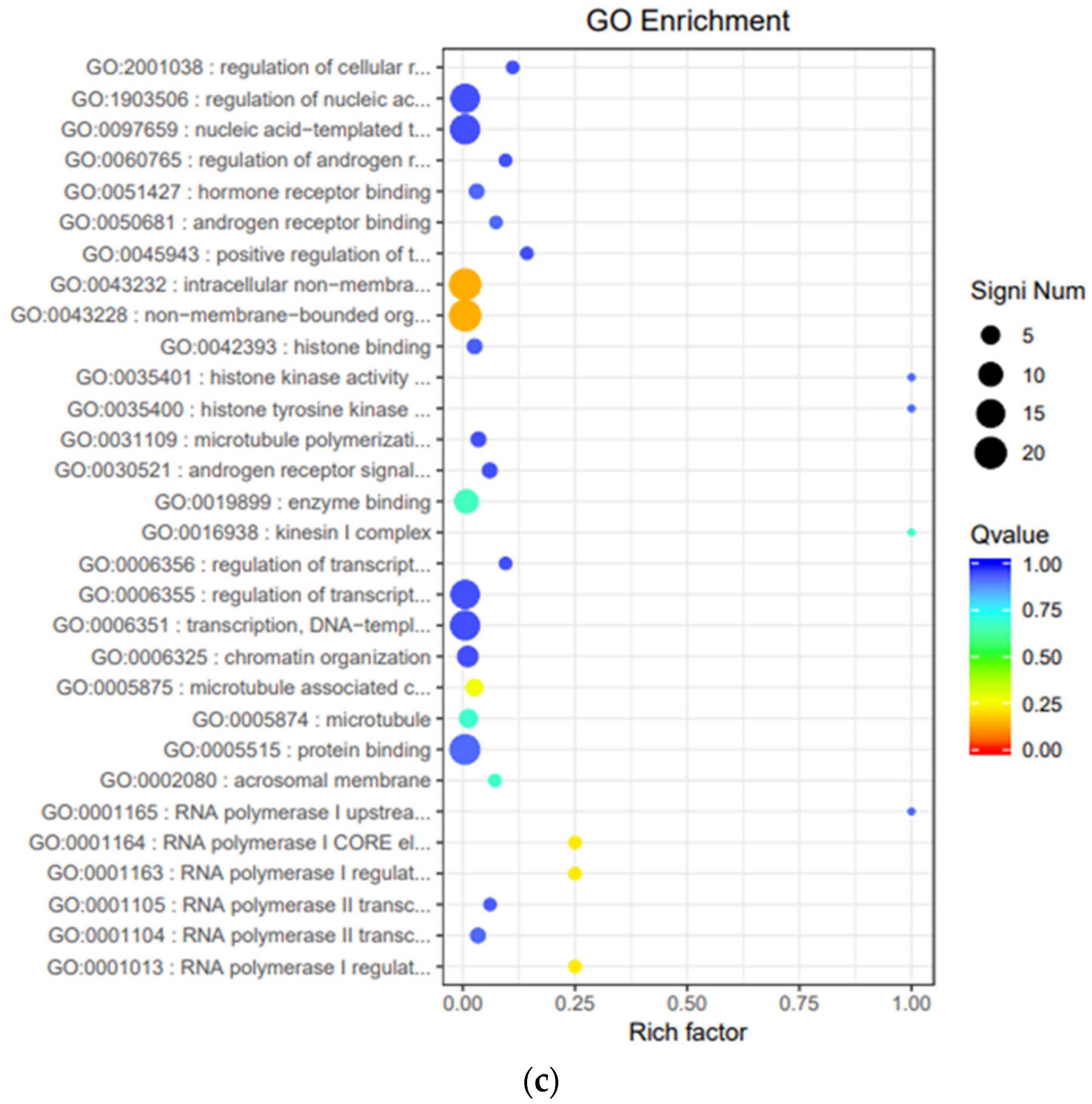
3.6. Functional Enrichment Analysis of Gene Ontology (GO)
3.7. KEGG Enrichment Analysis
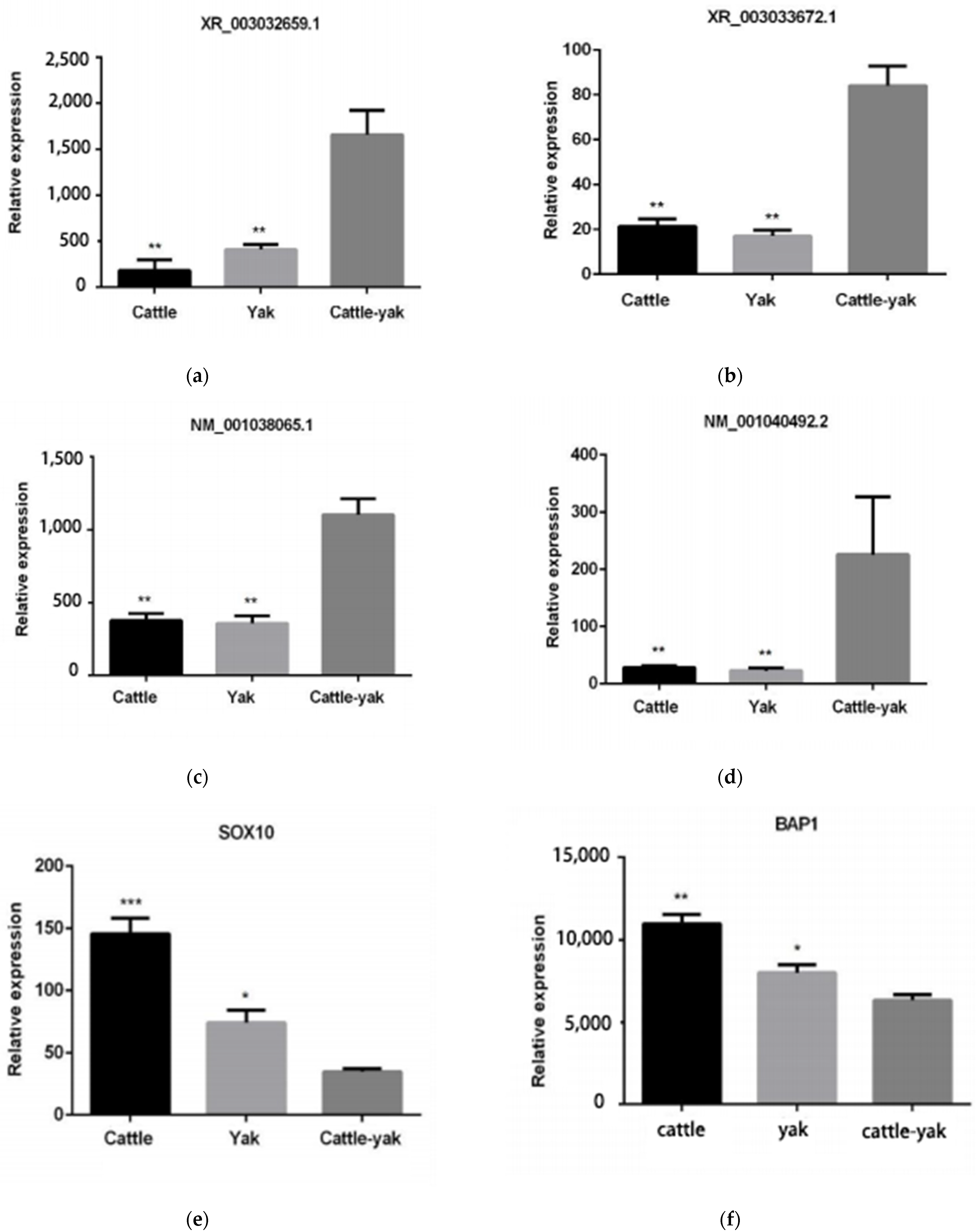
3.8. Validation of qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yan, P.; Xiang, L.; Guo, X.; Bao, P.-J.; Jin, S.; Wu, X.-Y. The low expression of Dmrt7 is associated with spermatogenic arrest in cattle-yak. Mol. Biol. Rep. 2014, 41, 7255–7263. [Google Scholar] [CrossRef]
- Tumennasan, K.; Tuya, T.; Hotta, Y.; Takase, H.; Speed, R.M.; Chandley, A.C. Fertility investigations in the F1 hybrid and backcross progeny of cattle (Bos taurus) and yak (B. grunniens) in Mongolia. Cytogenet. Cell Genet. 1997, 78, 69–73. [Google Scholar] [CrossRef]
- Zhao, W.; Ahmed, S.; Ahmed, S.; Yangliu, Y.; Wang, H.; Cai, X. Analysis of long non-coding RNAs in epididymis of cattleyak associated with male infertility. Theriogenology 2021, 160, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wu, S.; Zhao, W.; Mipam, T.; Liu, J.; Liu, W.; Yi, C.; Shah Ma Yu, S.; Cai, X. Differentially expressed microRNAs between cattleyak and yak testis. Sci. Rep. 2018, 8, 592. [Google Scholar] [CrossRef]
- Coyne, J.A. The genetic basis of Haldane’s rule. Nature 1985, 314, 736–738. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-C.; Wang, G.-W.; Xu, S.-R.; Zhang, X.-N.; Yang, Q.-E. The expression of histone methyltransferases and distribution of selected histone methylations in testes of yak and cattle-yak hybrid. Theriogenology 2020, 144, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.-L.; Song, H.-F.; Guan, J.-Q. Investigation on mechanism of sterility of male hybrids between yak and cattle. J. Appl. Anim. Res. 2014, 42, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Niayale, R.; Cui, Y.; Adzitey, F. Male hybrid sterility in the cattle-yak and other bovines: A review. Biol. Reprod. 2020, 104, 495–507. [Google Scholar] [CrossRef]
- Fedyk, S.; Krasińska, M. Studies on the spermatogenesis in European bison and domestic cattle hybrids. Acta Theriol. 1971, 16, 449–464. [Google Scholar] [CrossRef] [Green Version]
- Guo, A.P. Comparative research on chromosome in yak, yellow cattle and its hybrid. J. Genet. 1983, 10, 137–143. [Google Scholar]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; Li, Y.; Li, B.; Luo, H.; Xu, H.; Pan, Z.; Xie, Z.; Li, Q. Epigenetic Regulation of Bovine Spermatogenic Cell-Specific Gene Boule. PLoS ONE 2015, 10, e0128250. [Google Scholar] [CrossRef]
- Yen, P.H.; Chai, N.N.; Salido, E.C. The Human Autosomal Gene DAZLA: Testis Specificity and a Candidate for Male Infertility. Human Mol. Genet. 1996, 5, 2013–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Yu, S.; Cui, Y.; He, J.; Zhang, Q.; Sun, J.; Huang, Y.; Yang, X.; Cao, M.; Liao, B.; et al. Regulation by Hsp27/P53 in testis development and sperm apoptosis of male cattle (cattle-yak and yak). J. Cell. Physiol. 2019, 234, 650–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Diao, Y.; Qi, S.; Pan, X.; Wang, Q.; Xin, Y.; Cao, X.; Ruan, J.; Zhao, Z.; Luo, L.; et al. Phosphorylated Hsp27 activates ATM-dependent p53 signaling and mediates the resistance of MCF-7 cells to doxorubicin-induced apoptosis. Cell. Signal. 2013, 25, 1176–1185. [Google Scholar] [CrossRef]
- Cai, X.; Yu, S.; Mipam, T.; Yang, F.; Zhao, W.; Liu, W.; Cao, S.; Shen, L.; Zhao, F.; Sun, L.; et al. Comparative analysis of testis transcriptomes associated with male infertility in cattleyak. Theriogenology 2017, 88, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Mipam, T.; Xu, C.; Zhao, W.; Shah, M.A.; Yi, C.; Luo, H.; Cai, X.; Zhong, J. Testis transcriptome profiling identified genes involved in spermatogenic arrest of cattleyak. PLoS ONE 2020, 15, e0229503. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, T.; Ohta, T.; Bono, H. Experimental design-based functional mining and characterization of high-throughput sequencing data in the sequence read archive. PLoS ONE 2013, 8, e77910. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.A.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2009, 38, 1767–1771. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- StringTie assembles transcriptomes. Nat. Methods 2015, 12, 288. [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [Green Version]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar Gustavo, A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef]
- Li, A.; Zhang, J.; Zhou, Z. PLEK: A tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform. 2014, 15, 311. [Google Scholar] [CrossRef] [Green Version]
- Sang, J.; Wang, Z.; Li, M.; Cao, J.; Niu, G.; Xia, L.; Zou, D.; Wang, F.; Xu, X.; Han, X.; et al. ICG: A wiki-driven knowledgebase of internal control genes for RT-qPCR normalization. Nucleic Acids Res. 2017, 46, D121–D126. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Buxadera, A.; Palacios-Espinosa, A.; Espinosa-Villavicencio, J.L.; Guerra-Iglesias, D. Genotype Environment interactions for milk production traits in Holstein and crossbred Holstein-Zebu cattle populations estimated by a character state multibreed model. Livest. Sci. 2016, 191, 108–116. [Google Scholar] [CrossRef]
- Shan, J.; Cai, Z.; Zhang, Y.; Xu, H.; Rao, J.; Fan, Y.; Yang, J. The underlying pathway involved in inter-subspecific hybrid male sterility in rice. Genomics 2019, 111, 1447–1455. [Google Scholar] [CrossRef]
- Lustyk, D.; Kinský, S.; Ullrich, K.K.; Yancoskie, M.; Kašíková, L.; Gergelits, V.; Sedlacek, R.; Chan, Y.F.; Odenthal-Hesse, L.; Forejt, J.; et al. Genomic Structure of Hstx2 Modifier of Prdm9-Dependent Hybrid Male Sterility in Mice. Genetics 2019, 213, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Dedukh, D.; Majtánová, Z.; Marta, A.; Pšenička, M.; Kotusz, J.; Klíma, J.; Juchno, D.; Boron, A.; Janko, K. Parthenogenesis as a Solution to Hybrid Sterility: The Mechanistic Basis of Meiotic Distortions in Clonal and Sterile Hybrids. Genetics 2020, 215, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Marguerat, S.; Bähler, J. RNA-seq: From technology to biology. Cell. Mol. Life Sci. 2010, 67, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croucher, N.J.; Thomson, N.R. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010, 13, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Gao, L.; Xu, E.Y. LncRNA, a new component of expanding RNA-protein regulatory network important for animal sperm development. Semin. Cell Dev. Biol. 2016, 59, 110–117. [Google Scholar] [CrossRef]
- Yang, L.; Yi, K.; Wang, H.; Zhao, Y.; Xi, M. Comprehensive analysis of lncRNAs microarray profile and mRNA-lncRNA co-expression in oncogenic HPV-positive cervical cancer cell lines. Oncotarget 2016, 7, 49917–49929. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [Green Version]
- Radovic, S.M.; Starovlah, I.M.; Capo, I.; Miljkovic, D.; Nef, S.; Kostic, T.S.; Andric, S.A. Insulin/IGF1 signaling regulates the mitochondrial biogenesis markers in steroidogenic cells of prepubertal testis, but not ovary†. Biol. Reprod. 2018, 100, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Kjærner-Semb, E.; Ayllon, F.; Kleppe, L.; Sørhus, E.; Skaftnesmo, K.; Furmanek, T.; Segafredo, F.T.; Thorsen, A.; Fjelldal, P.G.; Hansen, T.; et al. Vgll3 and the Hippo pathway are regulated in Sertoli cells upon entry and during puberty in Atlantic salmon testis. Sci. Rep. 2018, 8, 1912. [Google Scholar] [CrossRef] [Green Version]
- Snider, L.P.; Snider, E.; Simmons, O.; Lilly, B.; Conway, S.J. Analysis of Uncharacterized mKiaa1211 Expression during Mouse Development and Cardiovascular Morphogenesis. J. Cardiovasc. Dev. Dis. 2019, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snider, P.L.; Simmons, O.; Conway, S.J. Cracd Marks the First Wave of Meiosis during Spermatogenesis and Is Mis-Expressed in Azoospermia Mice. J. Develop. Biol. 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Takahashi, K.; Niide, O.; Shibata, M.; Fukuzawa, M.; Ra, C. LDOC1, a novel MZF-1-interacting protein, induces apoptosis. FEBS Lett. 2005, 579, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Sane, S.; Rezvani, K. Essential Roles of E3 Ubiquitin Ligases in p53 Regulation. Int. J. Mol. Sci. 2017, 18, 442. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Total Reads | Total Map | Unique Map | Multiple Map | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|
| H1 | 66,835,092 | 62,423,261 | 59,139,082 (88.49%) | 1,561,762 (2.34%) | 95.14% | 50.09% |
| H2 | 66,125,724 | 61,820,573 | 58,508,258 (88.48%) | 1,627,394 (2.46%) | 95.37% | 50.19% |
| H3 | 72,510,220 | 67,528,071 | 63,952,926 (88.20%) | 1,727,552 (2.38%) | 95.18% | 50.49% |
| M1 | 72,889,560 | 64,684,443 | 60,624,382 (83.17%) | 1,808,268 (2.48%) | 95.02% | 50.45% |
| M2 | 67,418,572 | 59,919,256 | 55,651,456 (82.55%) | 1,701,518 (2.52%) | 95.17% | 49.84% |
| M3 | 55,624,916 | 48,314,822 | 43,426,696 (78.07%) | 1,083,154 (1.95%) | 95.02% | 50.57% |
| P1 | 68,746,398 | 60,688,223 | 56,257,718 (81.83%) | 1,662,678 (2.42%) | 95.04% | 51.28% |
| P2 | 71,810,310 | 64,010,649 | 59,921,584 (83.42%) | 1,673,008 (2.33%) | 95.13% | 50.64% |
| P3 | 85,334,762 | 76,830,344 | 71,423,888 (83.70%) | 2,686,240 (3.15%) | 94.67% | 51.22% |
| Class Code | Class Code Description | Number |
|---|---|---|
| = | Complete match of intron chain | 20,039 |
| C | Contained | 351 |
| J | Potentially novel isoform (fragment): at least one splice junction | 16,054 |
| E | Single exon transfrag overlapping a reference exon and 10 bp intron | 101 |
| I | A transfrag falling entirely within a reference intron | 6701 |
| O | Generic exonic overlap with a reference transcript | 845 |
| R | Repeat. Currently determined by looking at the soft-masked | 0 |
| U | Unknown, intergenic transcript | 10,893 |
| P | Possible polymerase run-on fragment | 725 |
| X | Exonic overlap with reference on the opposite strand | 3042 |
| S | An intron of the transfrag overlaps reference intron | 46 |
| Y | No exon overlap: ref exons falling within transfrag introns | 646 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.; Chen, T.; Luo, X.; Chen, S.; Wang, J.; Lai, S.; Jia, X. Identification of Novel lncRNA and Differentially Expressed Genes (DEGs) of Testicular Tissues among Cattle, Yak, and Cattle-Yak Associated with Male Infertility. Animals 2021, 11, 2420. https://doi.org/10.3390/ani11082420
Zhao S, Chen T, Luo X, Chen S, Wang J, Lai S, Jia X. Identification of Novel lncRNA and Differentially Expressed Genes (DEGs) of Testicular Tissues among Cattle, Yak, and Cattle-Yak Associated with Male Infertility. Animals. 2021; 11(8):2420. https://doi.org/10.3390/ani11082420
Chicago/Turabian StyleZhao, Shaokang, Tingting Chen, Xinmao Luo, Shiyi Chen, Jie Wang, Songjia Lai, and Xianbo Jia. 2021. "Identification of Novel lncRNA and Differentially Expressed Genes (DEGs) of Testicular Tissues among Cattle, Yak, and Cattle-Yak Associated with Male Infertility" Animals 11, no. 8: 2420. https://doi.org/10.3390/ani11082420
APA StyleZhao, S., Chen, T., Luo, X., Chen, S., Wang, J., Lai, S., & Jia, X. (2021). Identification of Novel lncRNA and Differentially Expressed Genes (DEGs) of Testicular Tissues among Cattle, Yak, and Cattle-Yak Associated with Male Infertility. Animals, 11(8), 2420. https://doi.org/10.3390/ani11082420