1. Introduction
In cattle, fetal death rates from fertilization to term can be up to 56 percent [
1]. More than 70 percent of these deaths occur during the first few months of pregnancy [
2]. Interestingly, most pregnancy losses occur during the first two to three weeks, when important physiological events, including blastocyst enlongation, pregnancy establishment and embryo implantation, are occurring in the endometrium [
1]. Recent studies have shown that the embryo may receive different inputs from the endometrium during its development, including embryotrophic factors (amino acids, carbohydrates, proteins, lipids and other substances) provided by the uterus [
1]. Moreover, endometrial gene expression patterns before and after the time of implantation (days 5–20) determine the ability of the uterine environment to maintain pregnancy [
3]. Therefore, the identification of genes responsible for the establishment and sustenance of pregnancy in cows could provide key information for the selection of informative genes responsible for increasing bovine fertility [
2]. Most reports have found differentially expressed genes in the first two to three weeks of pregnancy, including the insulin-like growth factor (
IGF) system [
1,
2,
4,
5], interleukin 1 (
IL1) [
6], claudin 10 (
CLND10), matrix Gla protein (MGP) [
7], connective tissue growth factor (
CTGF), solute carrier family 5 member 1 (
SLC5A1), lactotransferrin (
LTF) [
3,
8,
9], ubiquitin-like modifier (
ISG15), complement C1 (
C1), complement C4 (
C4), CXC motif chemokine ligand 5 (
CXCL5), alanyl aminopeptidase (
ANPEP), fatty acid binding protein 3 (
FABP3), lipoprotein lipase (
LPL), solute carrier family 2 member 5 (
SLC2A5) [
3,
9], semaphorin 3E (
SEMA3E), collagen type IV alpha 1 chain (
COL4A1) and phospholipase A2 (
PLA2) [
2]. It can be inferred that the identification of cause–effect relationships from differentially expressed genes in the uterus within 5 to 16 days of pregnancy may identify biomarkers and core genes responsible for successful and sustained pregnancy.
To date, several methods have been used to extract the network structure of gene expression data. These include linear regression, neural networks, differential equations, Boolean networks and Bayesian networks (BNs) [
10,
11]. Previous studies have shown that BNs perform better than the other methods for structural reconstruction. BNs are a class of graphical models connecting variables (nodes) by edges (arcs) [
10,
11]. These networks are examples of the application of graph theory and conditional probability rules for extracting network structures between existing variables of a dataset. BN learning methods are classified into parameter learning and structure learning [
12]. Different algorithms have already been proposed for the structure learning of BNs, including constraint-based algorithms (CBAs), score-based algorithms (SBAs), and hybrid algorithms (HAs) [
12]. CBA methods seek to estimate the structure of the BN using different conditional independence tests. In CBAs, it is not possible to fully extract the causal relationships between genes; thus, structures reconstructed this way are called partially directed acyclic graphs. CBA methods can be classified into grow-shrink (GS) and max-min parent children (MMPC) algorithms. GS algorithms are used for identifying a Markov blanket (MB) in a BN. MMPC algorithms use a forward-looking selection technique for identifying neighbors in a graph [
13]. On the other hand, SBAs are a group of heuristic optimization algorithms that find the best structure according to diverse predefined score functions. In the HA category, two types of algorithms, called maximum-minimum hill-climbing (MMHC) and restricted maximization (RSMAX), have been suggested to solve some limitations of the above-mentioned categories [
12]. MMHC uses the features of an MMPC algorithm (to limit search space) and hill-climbing (HC) to find the network with the highest rank in the restricted space [
12]. RSMAX is a flexible algorithm that is able to hybridize different characteristics of both the CBA and SBA categories [
12].
Given the different characteristics of the aforementioned algorithms but the similar goal (finding the best structure), it is acknowledged that the hub genes identified from each algorithm may be somewhat different. Extracting the important gene networks can help identify genes associated with complex diseases and traits, showing the associations between genes and generalizing the relationships and processes in which genes interact [
14]. Hub and regulatory genes in gene networks can be extracted using the above BN categories. Although the best algorithm might be assumed to be the one that hits the most gene annotations, a consensus of all the above BN categories can be taken as the most reliable. In addition, to date, there have not been any comparisons of the effectiveness of different BN algorithms in identifying hub genes in transcriptomic data, especially in cattle.
Therefore, the aims of the present study are to (1) identify hub genes in the bovine uterine transcriptome using two subclasses of the three main algorithms, including CBAs (GS and MMPC), SBAs (HC and TS), and HAs (MMHC and RSMAX); (2) detect hub genes shared between learned structures of the mentioned algorithms; and (3) find the best structural learning BN algorithm for the GSE33030 dataset based on biological classifications and annotations.
4. Discussion
Using a uterine transcriptomic dataset that examined the effects of progesterone during pregnancy in cattle, we used different modelling algorithms to identify core genes playing significant roles in gene expression networks. In order to create the relevant Bayesian networks (BNs), constraint-based algorithms (CBAs), score-based algorithms (SBAs) and hybrid algorithms (HAs) were all investigated. The SBAs identified the most network connections. The highest total number of connections and directed connections observed with SBAs was probably due to the complete reconstruction of the network structure and the determination of causal relationships between genes identified with these algorithms. Due to their inherent theory, the other two algorithms were not fully capable of determining the causal relationships between all the genes. Theoretically, the concept of the Markov Blanket (MB) refers to the parents, children and spouse of a gene [
13]. Extraction of an MB for small-scale networks was one of the network modeling parameters. The aim of the network extraction process by CBAs was to initially identify the MB. Another concept underlying BNs is that of the neighborhood size (NS), which refers to the adjacent genes of a particular gene, plus edges connecting these adjacent nodes, and is very useful in identifying modules in the network. The MB and NS are likely to indicate, in part, the different clusters and nodes in the studied gene expression series. Given the low estimated values for the tabu search (TS) and CBA factors, the probability of the formation of clusters in the GSE33030 dataset was very low. The branching factor (BF), representing the number of genes that can be affected by a specific gene, was one of the factors that created clusters in networks [
13]. The RSMAX algorithm from the HAs used a smaller number of conditional independence tests for structural reconstruction compared to the other algorithms.
One of the ways to compare different reconstructed networks is to use their global topological parameters, such as betweenness, eccentricity and degree [
14]. Network topology often shows information about the biological importance of a network. Topological parameters help to better recognize the consequences of the hub genes in a network. The degree of connectedness of a gene in a directed graph refers to the number of incoming and outgoing arcs, namely, in-degree and out-degree, respectively. The degrees of genes indicate one of the major topological properties used to identify hub nodes in a graph. We adopted the definition of hub genes as genes with high correlation in the candidate module and high connectivity, as well as required to meet the absolute values of gene module membership (>0.80) and gene trait significance (>0.20) [
24]. Interestingly, the reconstructed network obtained by the SBAs showed the highest degree values (7.2 and 7.0 for HC and TS, respectively) compared to the other two algorithms.
The clustering coefficient is a criterion for measuring the tendency of a graph to form consecutive clusters and shows a subset of genes that contains many connections to these genes. The closer the clustering value is to 1, the greater the probability of cluster formation in the gene network [
14]. Remarkably, the SBA methods reported the highest clustering coefficient values, showing a relatively high number of co-expressed genes in the network. On the whole among the six different methods, low values of cluster coefficients showed a stochastic structure, as previous studies have shown that biological networks do not show strong tendency to shape clusters [
14]. The lowest eccentricity values were obtained in graphs reconstructed by SBAs (3.5 and 3.5 for HC and TS, respectively), indicating high connectivity among genes in the reconstructed gene network. In fact, eccentricity shows the greatest distance between a particular node (‘gene’ in our context) and any other nodes in the graph [
14].
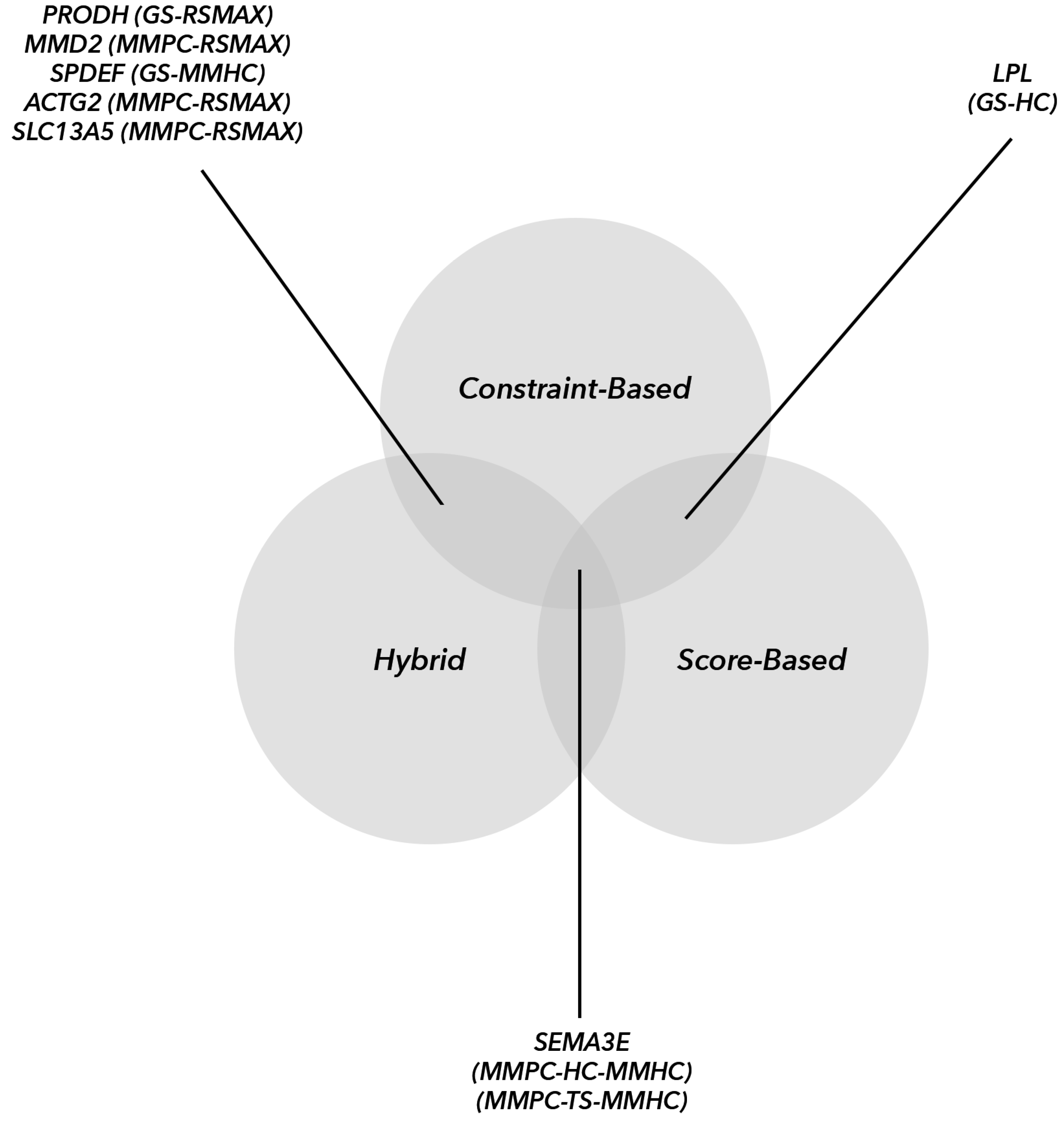
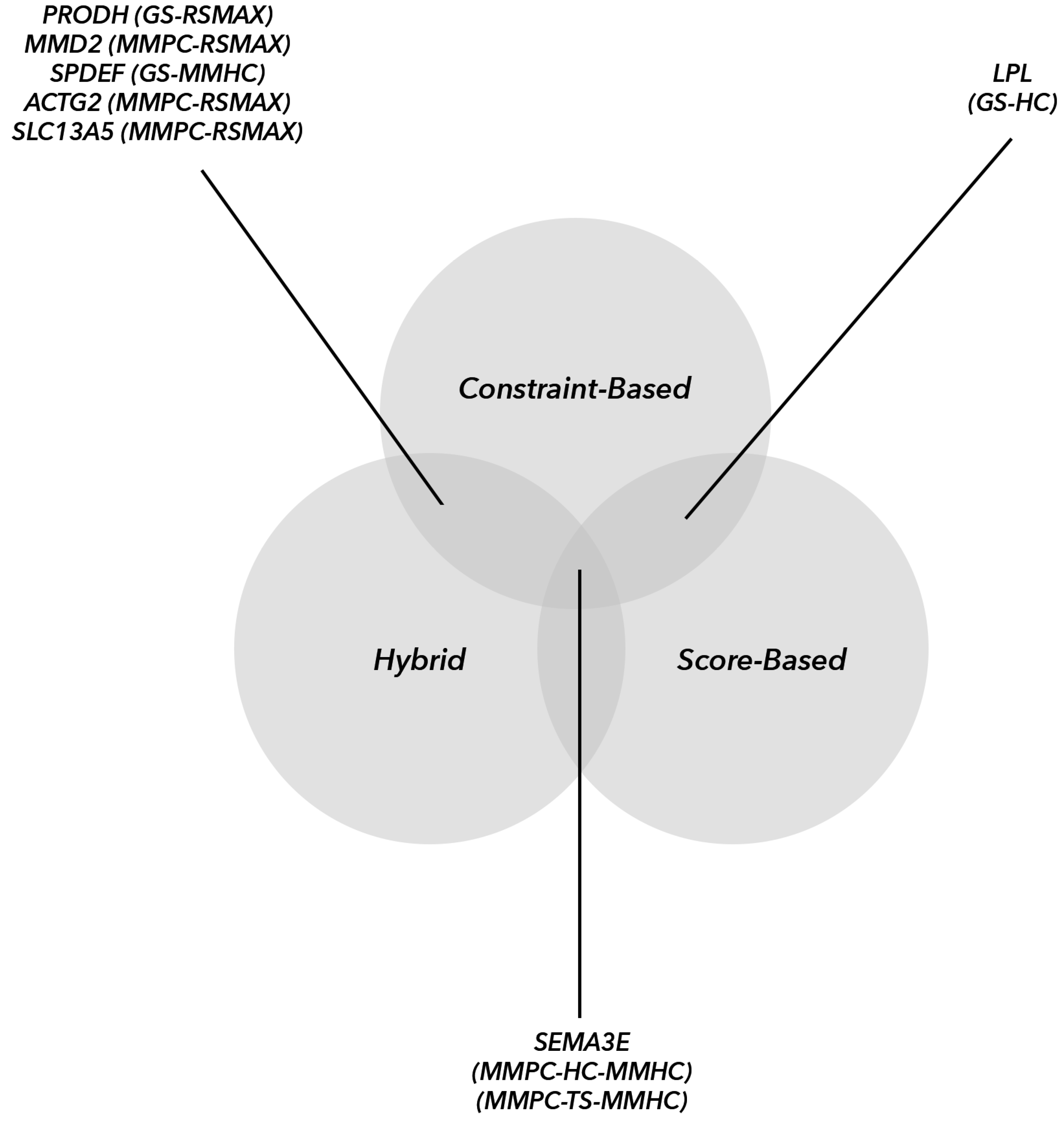
The higher number of commonly identified hub genes compared to the other two groups was probably due to common and identical reconstructions by the three main BN algorithmic groups. We recommend using other topological parameters as criteria to identify transcriptomic hub genes. Additionally, the
SEMA3E hub gene was shared among all three algorithmic groups (HAs, CBAs and SBAs). Research has shown that
ISG15 is a candidate gene for pregnancy recognition or return to the estrus cycle in cows [
9].
ISG15 is also known as a candidate gene for embryo implantation in the uterus. Diacylglycerol O-acyltransferase 2 (
DGAT2) is another hub gene identified with a biological role in pregnancy. It has eight exons, is found on chromosome 15 and is involved in lipid biosynthesis [
25]. This gene encodes one of two enzymes responsible for the catalytic reaction of the final step in triglyceride synthesis in which diacylglycerol attaches to long-chain fatty acyl-CoAs with a covalent bond. It has been shown through previous studies that the protein encoded by this gene is an enzyme involved in the synthesis of milk fat [
26] and is known as a marker and candidate gene in determining the fat content of milk [
25]. It was also reported that triglyceride is another potential energy source for the bovine blastocyst, and the
DGAT2 catalyst is the final stage in its synthesis [
27]. Progesterone appears to stimulate the expression of
DGAT2 in the endometrium [
7], and defects of intrauterine growth retardation due to
DGAT2 deficiency were observed, indicating that the presence of this enzyme is necessary for the development of uterine embryos [
28]. By regulating the expression of
DGAT2, progesterone stimulates blastocyst growth in the pre-implantation stage in the uterus [
29]. Progesterone injection into the uterus increased the expression of
DGAT2, which in turn triggered triglyceride synthesis reactions and the transfer of glucose in the uterus [
3]. Increasing the expression of
DGAT2 through progesterone led to the secretion of histotroph via the endometrium.
DGAT2 was recognized as a hub gene in our CBA analysis. However, other identified hub genes with no clear biological roles may be involved in metabolic or immune processes. The results in
Table 3 show that identification of hub genes by each algorithmic category was partly affected by the reconstructed graph, as well as by the implemented topological parameters.
A useful tool for investigating reproductive problems in livestock could be to examine the expression of these core network genes in different tissues. In this way, general hubs and tissue-specific hubs can be identified. In order to find general hub genes ranked high in a set of tissues, the rank product method could be used [
30]. We believe that MB can be used to mine the whole network for ranked genes by the number of neighbors in the gene network. A gene’s rank product is the product of its ranks from each network. For locating hubs specific to a group of tissues, rank product could be used to rank hubs in both the target group of tissues and all other tissues, separately.
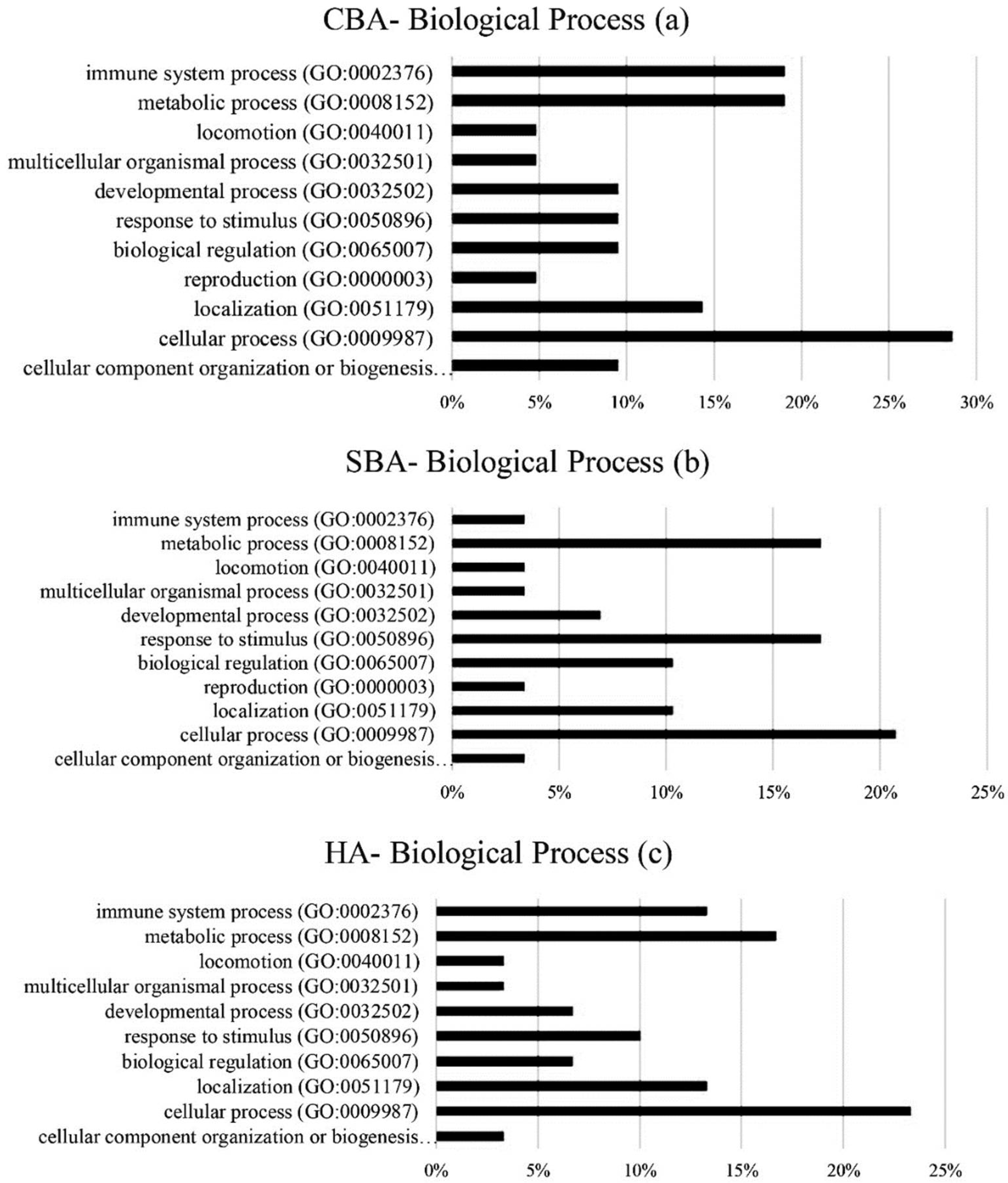
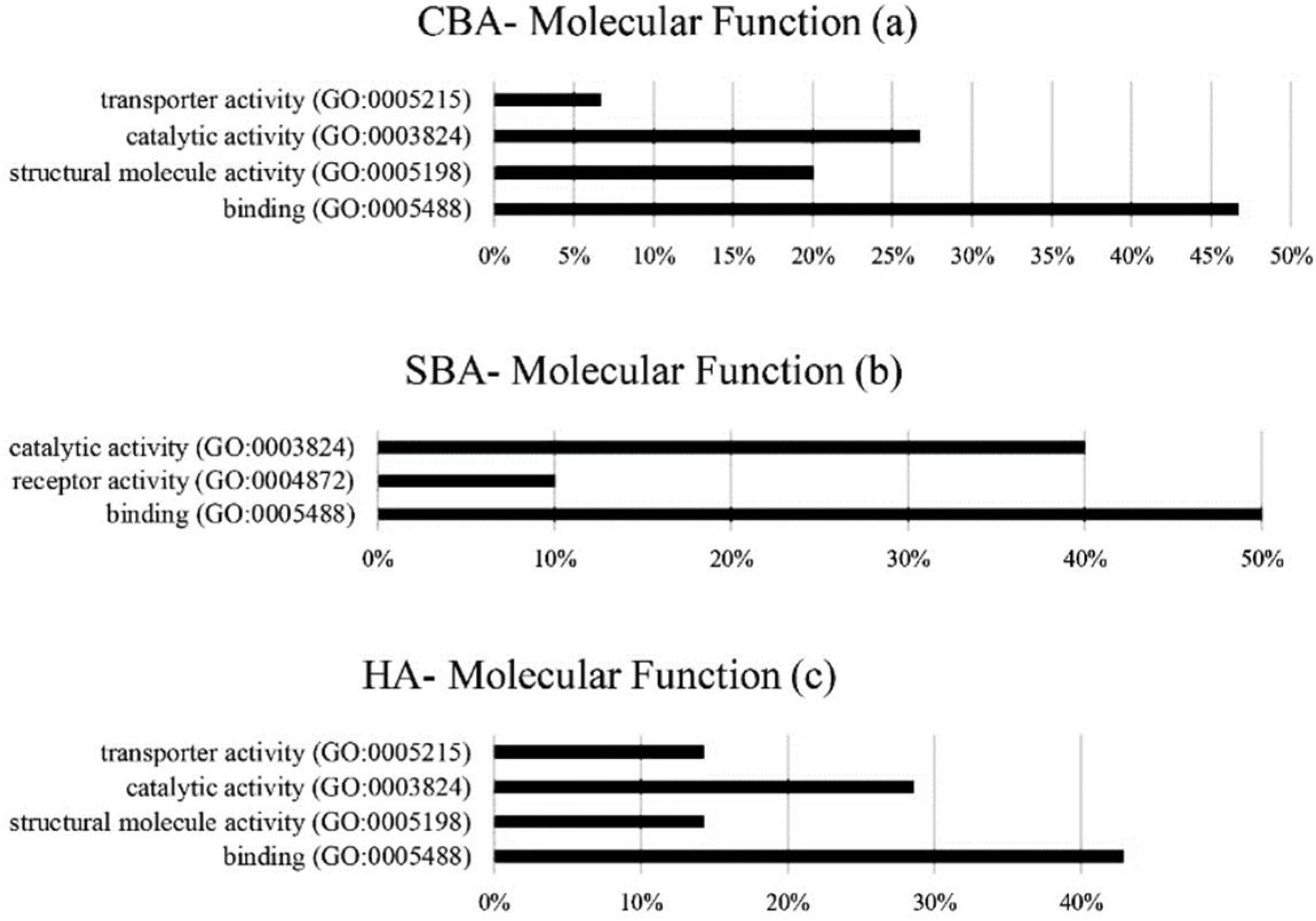
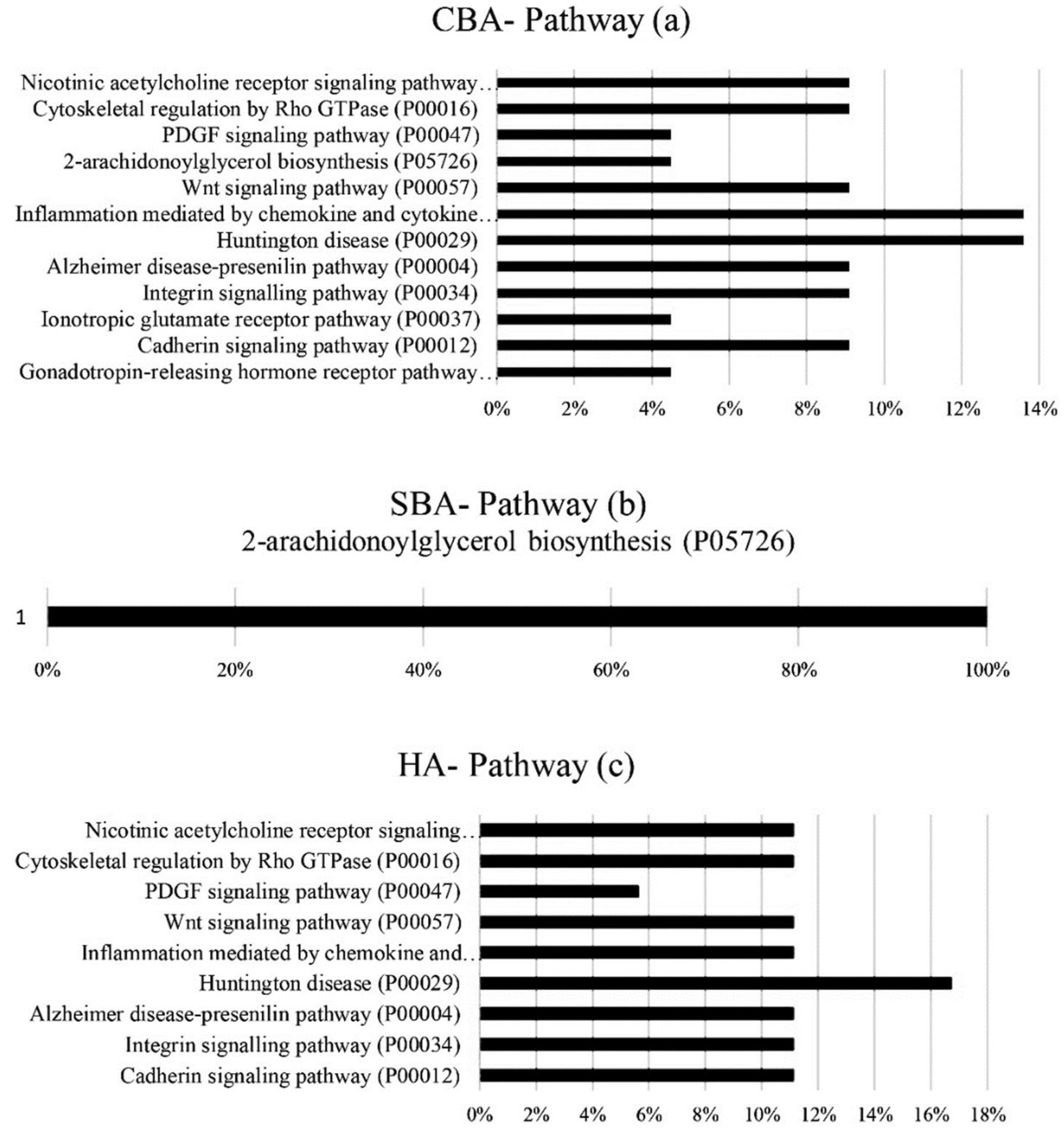
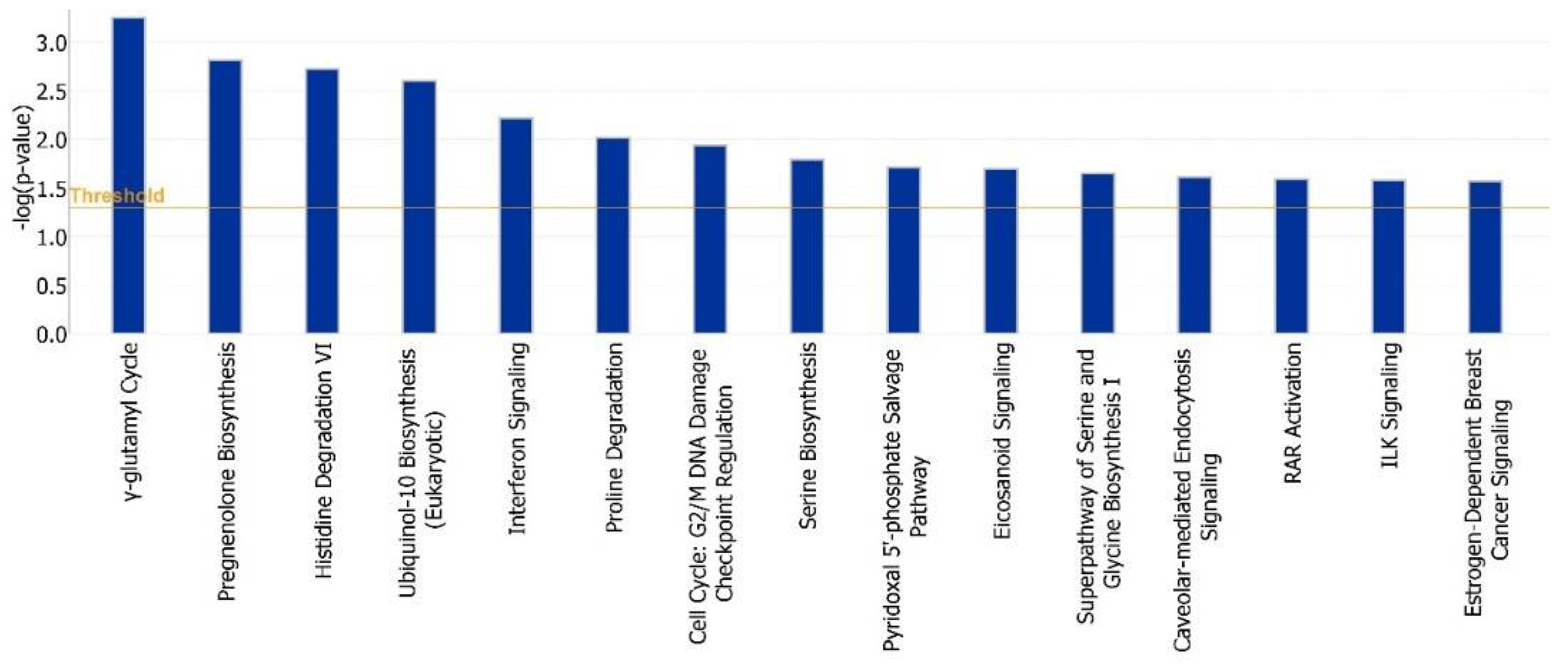
The hub genes derived from CBA methodology were involved in extracellular and alternative splicing pathways, with the annotation of hub genes from SBA analysis showing genes involved in extracellular pathways, secretion, glycerolipid metabolism and the formation of disulfide bonds [
31]. The annotation of the hub genes derived from the HA analysis also included genes involved in extracellular pathways, alternative splicing and the metabolism of arginine and proline [
31]. Based on the original GSE33030 dataset, high concentrations of progesterone secretion during the period of 7 to 16 days of pregnancy were conducive for embryo implantation in the uterus, indicating that during this stage, the pathways and genes relating to the production of hormones, enzymes and molecules related to the preservation and continuation of pregnancy were heavily activated. The production of growth hormones, binding molecules, chemokines and cytokines ensures the uterus is ready for embryo implantation [
32,
33]. The results of the hub gene annotation in this study also indicated that alternative splicing and extracellular pathways are important. The highest numbers of identified hub genes (7 to 8) that were identified by all the algorithms were in these pathways. Therefore, the pathways for the synthesis of hormones and enzymes were essential for preparing the uterus for implantation during days 5 to 13 of pregnancy. In cell biology, the extracellular space refers to gene products that exit from the plasmid membrane and flow through the intercellular fluids. Extracellular compounds include metabolites, ions, proteins and products such as RNA, DNA, lipids and microbial products, which affect endometrium function. Alternative splicing also plays an important role in the diversity of proteins derived from a particular transcript.
In the SBA analysis, the pathways for protease binding, disulfide bonds and glycerolipid metabolism were also significant. Isopeptide bonds also play a role in the binding of two amino acids forming polypeptides. Disulfide bonds interconnect between polypeptide units in proteins and form the tertiary structure of proteins, whereas the “protease bonds” category of genes are involved in protein decomposition. The metabolism of glycolipids is also essential for the synthesis of progesterone steroid hormones and other genes (DGAT2) in preparing the uterus for implantation (days 5 to 13). The hub gene annotation results showed that genes identified using SBAs and HAs were enriched for binding pathways (disulfide bands, isopeptide and protease), the production of enzymes and protein products (extracellular space and alternative splicing) and the synthesis of progesterone steroid hormone (glycerolipid metabolism). Therefore, considering the hub gene annotation results led to a more accurate and relevant prediction of the genes involved in the pathways of enzyme synthesis, the binding of polypeptide units and the synthesis of progesterone-related hormone during pregnancy establishment. In comparison to CBAs and HAs, the SBA methods found genes more related to the experimental treatment (in this case, the progesterone effect from days 5 to 16 of pregnancy in the GSE33030 microarray study). However, it is important to investigate the relevant biological and physiological functions of genes identified by all three algorithmic groups. These results classified hub genes based on their biological processes, molecular functions, cellular components, pathways and protein classes for further investigation. It was shown that models of gene regulation differed depending on the biological state of dairy cattle. Therefore, the importance of hub genes should be determined within the relevant biological context.
Cellular processes involve complicated cascades of biochemical reactions and signaling pathways. For correct cell function, these processes are required to be tightly controlled. Dysregulation of any element of these pathways can lead to a vast array of pathologies. By elevating progesterone during days 7 to 12 of estrus or pregnancy, a series of processes to synthesize and secrete progesterone for embryo implantation are activated, leading to the enriched gene expression of metabolic processes. During preimplantation, endometrial gene expression is regulated by the secretion of progesterone and interferon tau, and patterns of endometrium gene expression may be regulated only by progesterone and interferon, or by both [
34]. Embryo implantation is also seen as an inflammatory immune response [
6]. Interferon tau is a type I interferon that plays an antiviral, anticoagulant and immune-stimulating role. Interferon tau induces the expression of a number of genes in the endometrium that are essential for the transfer of food to the embryo or increase the expression of genes necessary to prepare the endometrium for implantation and continued pregnancy. Most of the hub genes identified from the CBA and HA analyses participated in immune system processes.
The results of this study were based on the use of bovine Affymetrix microarrays. The biological validation of the identified genes may be influenced by data type with respect to using microarrays versus RNA-Seq [
6]. Although a limitation of this study was the lack of another independent dataset in the database for validation of the hub genes, the goal of comparing three common algorithms for the detection of hub genes shared between learned structures of the algorithms was achieved. RNA-Seq allows full sequencing of the whole transcriptome, while microarrays only profile predefined transcripts and genes through hybridization. The ability of RNA-Seq to identify more differentially modulated transcripts of biological relevance, splice variants and non-coding transcripts, such as microRNAs, long non-coding RNAs and pseudogenes, makes it superior to microarrays. This difference has additional implications for mechanistic investigations or biomarker discovery [
6] making RNA-Seq data more useful with higher predictive power [
6].


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}