
Weighted Single-Step Genomic Best Linear Unbiased Prediction Method Application for Assessing Pigs on Meat Productivity and Reproduction Traits

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotypes
2.2. Genotypes
2.3. Statistical Analysis and Assessment
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, L.R. Strategy for applying genome-wide selection in dairy cattle. J. Anim. Breed. Genet. 2006, 123, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Ardestani, S.S.; Jafarikia, M.; Sargolzaei, M.; Sullivan, B.; Miar, Y. Genomic Prediction of Average Daily Gain, Back-Fat Thickness, and Loin Muscle Depth Using Different Genomic Tools in Canadian Swine Populations. Front. Genet. 2021, 12, 735. [Google Scholar] [CrossRef]
- Misztal, I. Inexpensive Computation of the Inverse of the Genomic Relationship Matrix in Populations with Small Effective Population Size. Genetics 2015, 202, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Pocrnic, I.; Lourenco, D.A.L.; Masuda, Y.; Legarra, A.; Misztal, I. The Dimensionality of Genomic Information and Its Effect on Genomic Prediction. Genetics 2016, 203, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Badke, Y.M.; Bates, R.O.; Ernst, C.W.; Fix, J.; Steibel, J.P. Accuracy of Estimation of Genomic Breeding Values in Pigs Using Low-Density Genotypes and Imputation. G3-Genes Genomes Genet. 2014, 4, 623–631. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, I.M.; Hayes, B.; Goddard, M. The Effects of Demography and Long-Term Selection on the Accuracy of Genomic Prediction with Sequence Data. Genetics 2014, 198, 1671–1684. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Enciso, M.; Rincón, J.C.; Legarra, A. Sequence- vs. chip-assisted genomic selection: Accurate biological information is advised. Genet. Sel. Evol. 2015, 47, 43. [Google Scholar] [CrossRef] [Green Version]
- Mrode, R.A. Linear Models for the Prediction of Animal Breeding Values, 3rd ed.; CABI: Boston, MA, USA, 2013. [Google Scholar]
- Lourenco, D.A.L. Introduction to Genomic Selection. University of Chicago: Piracicaba, Brazil, 2019. Available online: http://nce.ads.uga.edu/wiki/lib/exe/fetch.php?media=gs_lourenco_2019a.pdf (accessed on 29 June 2022).
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet. Res. 2012, 94, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Braz, C.U.; Taylor, J.F.; Bresolin, T.; Espigolan, R.; Feitosa, F.L.B.; Carvalheiro, R.; Baldi, F.; De Albuquerque, L.G.; De Oliveira, H.N. Sliding window haplotype approaches overcome single SNP analysis limitations in identifying genes for meat tenderness in Nelore cattle. BMC Genet. 2019, 20, 8. [Google Scholar] [CrossRef]
- Cherel, P.; Pires, J.; Glénisson, J.; Milan, D.; Iannuccelli, N.; Hérault, F.; Damon, M.; Le Roy, P. Joint analysis of quantitative trait loci and major-effect causative mutations affecting meat quality and carcass composition traits in pigs. BMC Genet. 2011, 12, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontanesi, L.; Schiavo, G.; Galimberti, G.; Calò, D.G.; Scotti, E.; Martelli, P.L.; Buttazzoni, L.; Casadio, R.; Russo, V. A genome wide association study for backfat thickness in Italian Large White pigs highlights new regions affecting fat deposition including neuronal genes. BMC Genom. 2012, 13, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, E.J.; Park, H.B.; Lee, J.B.; Yoo, C.K.; Kim, B.M.; Kim, H.I.; Kim, B.W.; Lim, H.T. Genome-wide association analysis identifies quantitative trait loci for growth in a Landrace purebred population. Anim. Genet. 2014, 45, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.-P.; Tribout, T.; Iannuccelli, N.; Bouffaud, M.; Servin, B.; Tenghe, A.; Dehais, P.; Muller, N.; Del Schneider, M.P.; Mercat, M.-J.; et al. A genome-wide association study of production traits in a commercial population of Large White pigs: Evidence of haplotypes affecting meat quality. Genet. Sel. Evol. 2014, 46, 12. [Google Scholar] [CrossRef] [Green Version]
- Strucken, E.M.; Schmitt, A.O.; Bergfeld, U.; Jurke, I.; Reissmann, M.; Brockmann, G.A. Genome wide study and validation of markers associated with production traits in German Landrace boars. J. Anim. Sci. 2014, 92, 1939–1944. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, A.; Kim, Y.-S.; Kang, J.-M.; Lee, Y.-M.; Rai, R.; Jung, J.-H.; Oh, D.-Y.; Nam, K.-C.; Lee, H.-K.; Kim, J.-J. Genome-wide Association Study to Identify Quantitative Trait Loci for Meat and Carcass Quality Traits in Berkshire. Asian-Australas. J. Anim. Sci. 2015, 28, 1537–1544. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, T.; Hayes, B.; Goddard, M. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Huang, Y.; Hou, L.; Ma, J.; Chen, C.; Ai, H.; Huang, L.; Ren, J. Genome-wide detection of genetic markers associated with growth and fatness in four pig populations using four approaches. Genet. Sel. Evol. 2017, 49, 21. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Liu, X.; Max, R.; Zhang, Z.; Zhao, S.; Fan, B. Genome-wide association study of the backfat thickness trait in two pig populations. Front. Agric. Sci. Eng. 2014, 1, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, M.C.; Zappaterra, M.; Davoli, R.; Zambonelli, P. Genome-wide association study identifies markers associated with carcass and meat quality traits in Italian Large White pigs. Anim. Genet. 2020, 51, 950–952. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, Z.; Glória, L.S.; Zhang, K.; Zhang, C.; Yang, R.; Luo, X.; Jia, X.; Lai, S.-J.; Chen, S.-Y. Genome-Wide Association Studies for Growth Curves in Meat Rabbits Through the Single-Step Nonlinear Mixed Model. Front. Genet. 2021, 12, 750939. [Google Scholar] [CrossRef] [PubMed]
- Fragomeni, B.O.; Lourenco, D.A.L.; Masuda, Y.; Legarra, A.; Misztal, I. Incorporation of causative quantitative trait nucleotides in single-step GBLUP. Genet. Sel. Evol. 2017, 49, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, D.; Misztal, I.; Tsuruta, S.; Aguilar, I.; Ezra, E.; Ron, M.; Shirak, A.; Weller, J. Methods for genomic evaluation of a relatively small genotyped dairy population and effect of genotyped cow information in multiparity analyses. J. Dairy Sci. 2014, 97, 1742–1752. [Google Scholar] [CrossRef] [Green Version]
- Veerkamp, R.F.; Bouwman, A.C.; Schrooten, C.; Calus, M.P.L. Genomic prediction using preselected DNA variants from a GWAS with whole-genome sequence data in Holstein–Friesian cattle. Genet. Sel. Evol. 2016, 48, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossi, D.A.; Brito, L.F.; Jafarikia, M.; Schenkel, F.S.; Feng, Z. Genotype imputation from various low-density SNP panels and its impact on accuracy of genomic breeding values in pigs. Animal 2018, 12, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Moghaddar, N.; Khansefid, M.; Van Der Werf, J.H.J.; Bolormaa, S.; Duijvesteijn, N.; Clark, S.A.; Swan, A.A.; Daetwyler, H.D.; MacLeod, I.M. Genomic prediction based on selected variants from imputed whole-genome sequence data in Australian sheep populations. Genet. Sel. Evol. 2019, 51, 72. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Liu, Y.; Yu, X.; Li, Y.; Yang, Y.; Wei, M.; Zhou, Q.; Wang, J.; Zhang, Y.; Zheng, W.; et al. Prediction of genomic breeding values based on pre-selected SNPs using ssGBLUP, WssGBLUP and BayesB for Edwardsiellosis resistance in Japanese flounder. Genet. Sel. Evol. 2020, 52, 49. [Google Scholar] [CrossRef]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. Plink: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Sargolzaei, M.; Iwaisaki, H.; Colleau, J.-J. A fast algorithm for computing inbreeding coefficients in large populations. J. Anim. Breed. Genet. 2005, 122, 325–331. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-Terwengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misztal, I.; Tsuruta, S.; Lourenco, D.; Masuda, Y.; Aguilar, I.; Legarra, A.; Vitezica, Z. BLUPF90 Family of Programs. University of Georgia: Athens, GA, USA, 2018. Available online: http://nce.ads.uga.edu/wiki/doku.php (accessed on 27 April 2022).
- Aguilar, I.; Misztal, I.; Johnson, D.L.; Legarra, A.; Tsuruta, S.; Lawlor, T.J. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- VanRaden, P. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, D.; Legarra, A.; Tsuruta, S.; Masuda, Y.; Aguilar, I.; Misztal, I. Single-Step Genomic Evaluations from Theory to Practice: Using SNP Chips and Sequence Data in BLUPF90. Genes 2020, 11, 790. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, E.; Kabanov, A.; Nikitin, S.; Somova, M.; Kharitonov, S.; Otradnov, P.; Kostyunina, O.; Karpushkina, T.; Martynova, E.; Sermyagin, A.; et al. Application of Genomic Data for Reliability Improvement of Pig Breeding Value Estimates. Animals 2021, 11, 1557. [Google Scholar] [CrossRef]
- Atashi, H.; Salavati, M.; De Koster, J.; Crowe, M.A.; Opsomer, G.; Hostens, M. The GplusE Consortium A Genome-Wide Association Study for Calving Interval in Holstein Dairy Cows Using Weighted Single-Step Genomic BLUP Approach. Animals 2020, 10, 500. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, D.A.L.; Fragomeni, B.O.; Bradford, H.L.; Menezes, I.R.; Ferraz, J.B.S.; Aguilar, I.; Tsuruta, S.; Misztal, I. Implications of SNP weighting on single-step genomic predictions for different reference population sizes. J. Anim. Breed. Genet. 2017, 134, 463–471. [Google Scholar] [CrossRef]
- Mehrban, H.; Naserkheil, M.; Lee, D.; Cho, C.; Choi, T.; Park, M.; Ibáñez-Escriche, N. Genomic Prediction Using Alternative Strategies of Weighted Single-Step Genomic BLUP for Yearling Weight and Carcass Traits in Hanwoo Beef Cattle. Genes 2021, 12, 266. [Google Scholar] [CrossRef]
- Teissier, M.; Larroque, H.; Robert-Granie, C. Accuracy of genomic evaluation with weighted single-step genomic best linear unbiased prediction for milk production traits, udder type traits, and somatic cell scores in French dairy goats. J. Dairy Sci. 2019, 102, 3142–3154. [Google Scholar] [CrossRef] [Green Version]
- Teissier, M.; Larroque, H.; Robert-Granié, C. Weighted single-step genomic BLUP improves accuracy of genomic breeding values for protein content in French dairy goats: A quantitative trait influenced by a major gene. Genet. Sel. Evol. 2018, 50, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lourenco, D.; Aguilar, I.; Legarra, A.; Misztal, I. Weighting Strategies for Single-Step Genomic BLUP: An Iterative Approach for Accurate Calculation of GEBV and GWAS. Front. Genet. 2016, 7, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarenga, A.; Veroneze, R.; De Oliveira, H.R.; Marques, D.B.D.; Lopes, P.S.; Silva, F.F.; Brito, L.F. Comparing Alternative Single-Step GBLUP Approaches and Training Population Designs for Genomic Evaluation of Crossbred Animals. Front. Genet. 2020, 11, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
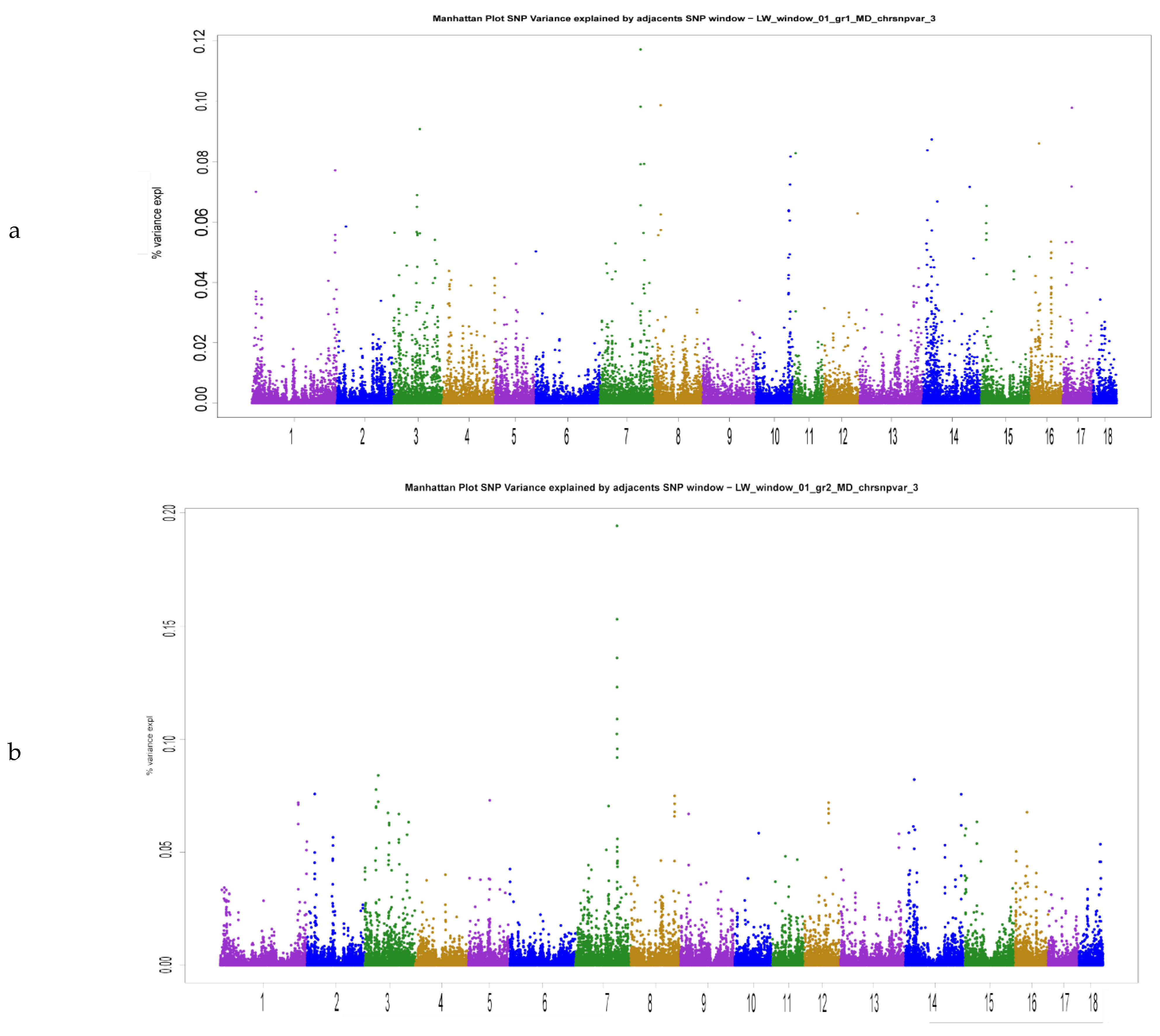
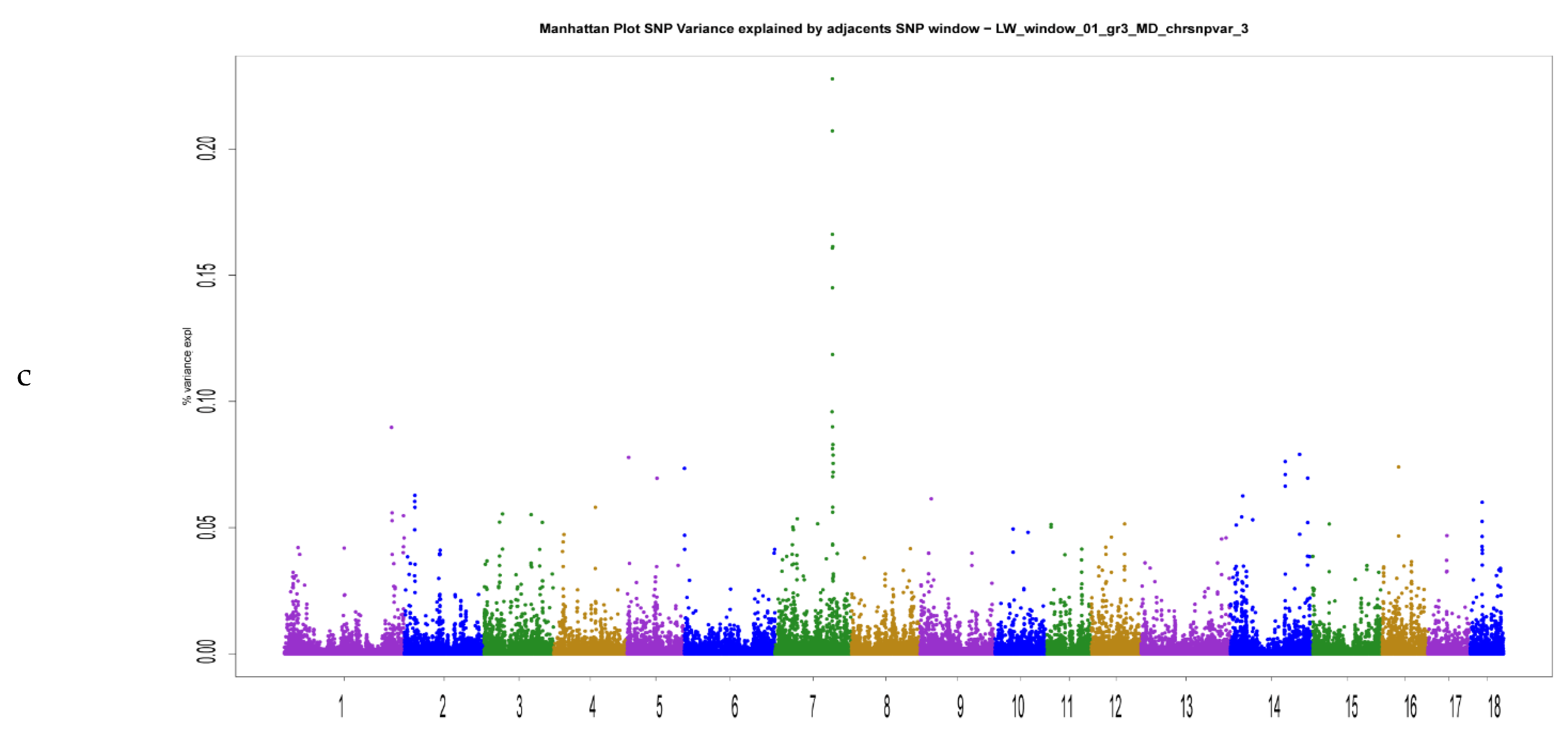
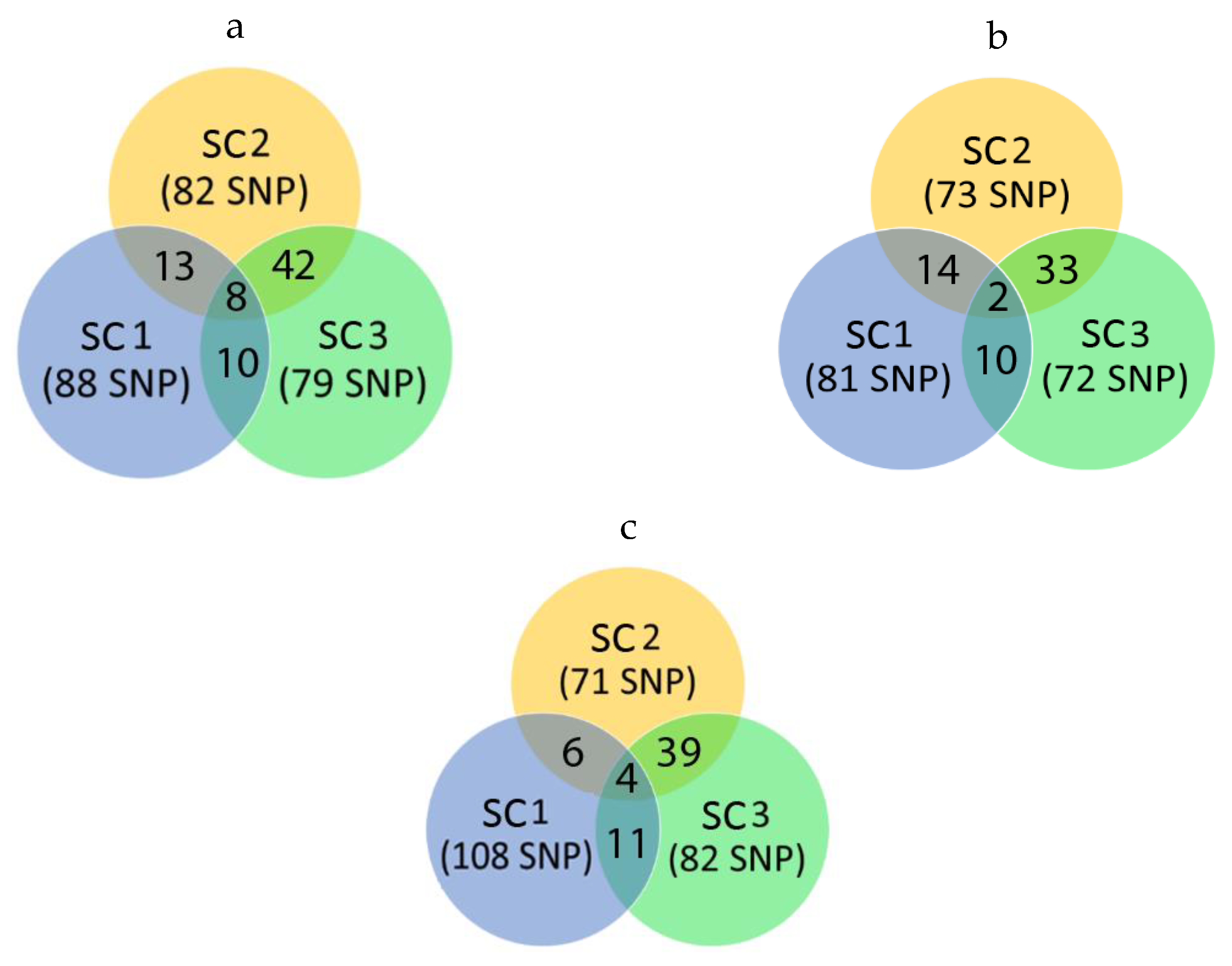


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean | SD | Range | h2 | N |
|---|---|---|---|---|---|
| BF1 | 16.13 | 3.63 | 7.0 to 34.0 | 0.428 | 62,927 |
| MD | 59.52 | 7.18 | 40.0 to 99.0 | 0.195 | |
| Age | 154.4 | 9.17 | 109 to 205 | 0.323 | |
| TNB | 15.4 | 4.14 | 1 to 29 | 0.119 | 16,070 |
| NBA | 14.2 | 3.95 | 0 to 28 | 0.112 |
| Scenario | Methods | Phenotypes and Pedigree Data | Genomic Data | Results |
|---|---|---|---|---|
| Scenario 1 | ssGBLUP wssGBLUP | WD | RG1 (RG4) | GEBVss1/4 GEBVwss1/4 |
| Scenario 2 | ssGBLUP wssGBLUP | WD | RG2 (RG5) | GEBVss2/5 GEBVwss2/5 |
| Scenario 3 | ssGBLUP wssGBLUP | WD | RG3 (RG6) | GEBVss3/6 GEBVwss3/6 |
| Scenario 4 | BLUP AM | WD | - | EBVWD |
| Scenario 5 (validation) | BLUP AM | - | PAPD | |
| ssGBLUP | PD | RG3 (RG6) | GEBVssPD | |
| wssGBLUP | RG3 (RG6) | GEBVwssPD |
| Parameter | Meat and Fattening Traits | ||
|---|---|---|---|
| Group 1 | Group 2 | Group 3 | |
| N | 530 | 1178 | 1493 |
| Ne | 85 | 90 | 96 |
| Reproduction traits | |||
| Group 4 | Group 5 | Group 6 | |
| N | 396 | 870 | 1228 |
| Ne | 96 | 107 | 107 |
| Trait | BLUP AM | ssGBLUP | wssGBLUP | ||||
|---|---|---|---|---|---|---|---|
| Meat and fattening traits | |||||||
| Scenario 4 | Scenario 1 | Scenario 2 | Scenario 3 | Scenario 1 | Scenario 2 | Scenario 3 | |
| BF1 | 0.723 | 0.716 | 0.757 | 0.767 | 0.919 | 0.931 | 0.933 |
| MD | 0.597 | 0.606 | 0.654 | 0.666 | 0.931 | 0.942 | 0.944 |
| Age | 0.679 | 0.675 | 0.718 | 0.728 | 0.983 | 0.986 | 0.986 |
| Reproduction traits | |||||||
| NBA | 0.438 | 0.465 | 0.484 | 0.499 | 0.803 | 0.803 | 0.813 |
| TNB | 0.441 | 0.473 | 0.492 | 0.511 | 0.826 | 0.828 | 0.840 |
| Correlation between | Trait | |||||
|---|---|---|---|---|---|---|
| BF1 | MD | Age | TNB | NBA | ||
| EBVWD | PAPD | 0.358 | 0.336 | 0.387 | 0.787 | 0.767 |
| GEBVssPD | 0.509 | 0.500 | 0.452 | 0.697 | 0.690 | |
| GEBVwssPD | 0.505 | 0.436 | 0.426 | 0.618 | 0.594 | |
| GEBVssWD | PAPD | 0.624 | 0.506 | 0.699 | 0.751 | 0.721 |
| GEBVssPD | 0.993 | 0.942 | 0.969 | 0.875 | 0.878 | |
| GEBVwssPD | 0.569 | 0.520 | 0.488 | 0.799 | 0.791 | |
| GEBVwssWD | PAPD | 0.356 | 0.278 | 0.373 | 0.615 | 0.574 |
| GEBVssPD | 0.565 | 0.498 | 0.479 | 0.686 | 0.693 | |
| GEBVwssPD | 0.562 | 0.457 | 0.466 | 0.713 | 0.716 | |
| EBV by Trait | GEBVss | GEBVwss | ||||
|---|---|---|---|---|---|---|
| Meat and fattening traits | ||||||
| Scenario 4 | Scenario 1 | Scenario 2 | Scenario 3 | Scenario 1 | Scenario 2 | Scenario 3 |
| EBV(BF1) | 0.974 | 0.950 | 0.945 | 0.977 | 0.956 | 0.951 |
| EBV(MD) | 0.957 | 0.927 | 0.910 | 0.926 | 0.918 | 0.911 |
| EBV(Age) | 0.961 | 0.937 | 0.931 | 0.965 | 0.948 | 0.944 |
| Reproduction traits | ||||||
| EBV(NBA) | 0.944 | 0.781 | 0.823 | 0.792 | 0.843 | 0.841 |
| EBV(TNB) | 0.944 | 0.769 | 0.808 | 0.775 | 0.840 | 0.846 |
| Trait/Group | Part of Variance Explained by Each SNP Marker | p-Value * | SNP Marker «Weight» | ||||
|---|---|---|---|---|---|---|---|
| Meat and fattening traits | |||||||
| Scenario 1 | Scenario 2 | Scenario 1 | Scenario 2 | Scenario 1 | Scenario 2 | ||
| BF1 | Scenario 2 | 0.429 | - | 0.382 | - | 0.408 | - |
| Scenario 3 | 0.352 | 0.850 | 0.314 | 0.834 | 0.352 | 0.839 | |
| MD | Scenario 2 | 0.511 | - | 0.454 | - | 0.479 | - |
| Scenario 3 | 0.435 | 0.816 | 0.353 | 0.790 | 0.390 | 0.796 | |
| Age | Scenario 2 | 0.394 | - | 0.366 | - | 0.404 | - |
| Scenario 3 | 0.335 | 0.822 | 0.291 | 0.809 | 0.330 | 0.811 | |
| Reproduction traits | |||||||
| NBA | Scenario 2 | 0.424 | - | 0.395 | - | 0.436 | - |
| Scenario 3 | 0.318 | 0.720 | 0.277 | 0.714 | 0.331 | 0.732 | |
| TNB | Scenario 2 | 0.420 | - | 0.384 | - | 0.423 | - |
| Scenario 3 | 0.294 | 0.725 | 0.263 | 0.716 | 0.313 | 0.733 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabanov, A.; Melnikova, E.; Nikitin, S.; Somova, M.; Fomenko, O.; Volkova, V.; Kostyunina, O.; Karpushkina, T.; Martynova, E.; Trebunskikh, E. Weighted Single-Step Genomic Best Linear Unbiased Prediction Method Application for Assessing Pigs on Meat Productivity and Reproduction Traits. Animals 2022, 12, 1693. https://doi.org/10.3390/ani12131693
Kabanov A, Melnikova E, Nikitin S, Somova M, Fomenko O, Volkova V, Kostyunina O, Karpushkina T, Martynova E, Trebunskikh E. Weighted Single-Step Genomic Best Linear Unbiased Prediction Method Application for Assessing Pigs on Meat Productivity and Reproduction Traits. Animals. 2022; 12(13):1693. https://doi.org/10.3390/ani12131693
Chicago/Turabian StyleKabanov, Artem, Ekaterina Melnikova, Sergey Nikitin, Maria Somova, Oleg Fomenko, Valeria Volkova, Olga Kostyunina, Tatiana Karpushkina, Elena Martynova, and Elena Trebunskikh. 2022. "Weighted Single-Step Genomic Best Linear Unbiased Prediction Method Application for Assessing Pigs on Meat Productivity and Reproduction Traits" Animals 12, no. 13: 1693. https://doi.org/10.3390/ani12131693