
Insight into the Fecal Microbiota Signature Associated with Growth Specificity in Korean Jindo Dogs Using 16S rRNA Sequencing
 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Extracting Microbial DNA and 16S rRNA Sequencing
2.3. Bioinformatics Analysis
2.4. LEfSe and Co-Abundance Network Analysis
2.5. Functional Prediction of Microbial Gene
3. Results
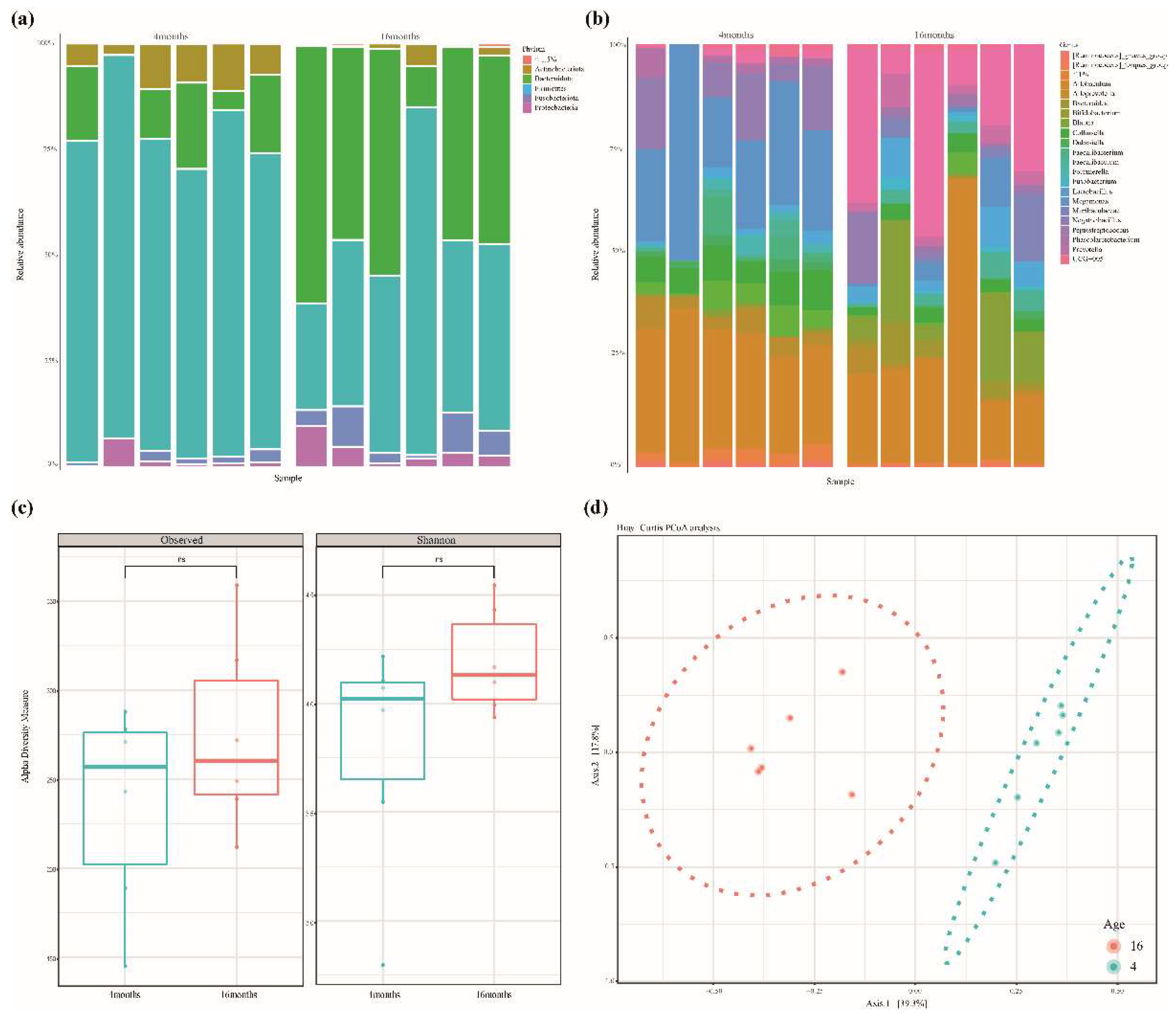
3.1. Sequencing Results Summary and Bacterial Structure
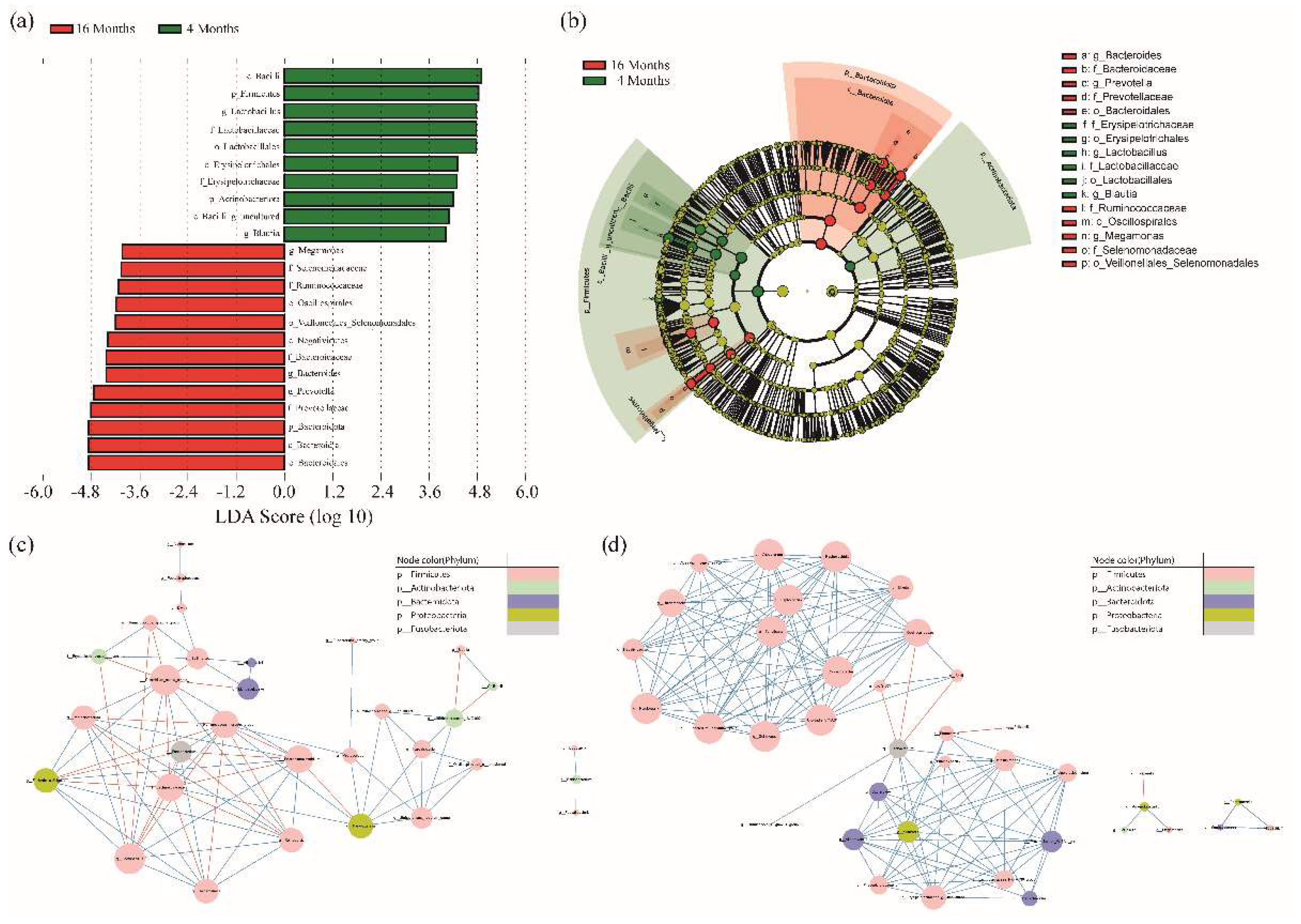
3.2. LEfSe Analysis and Co-Occurrence Analysis among the Microbial Genera
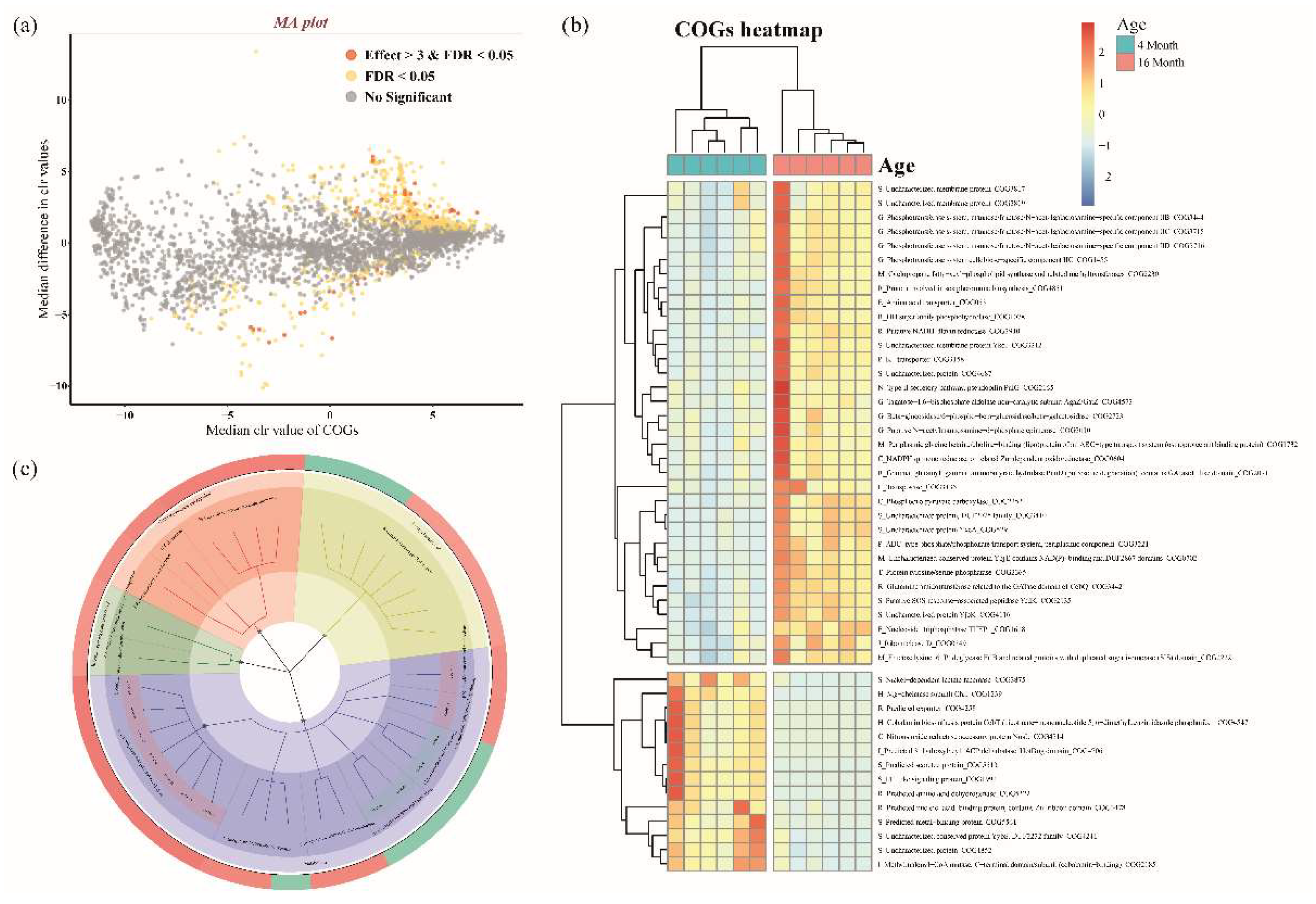
3.3. Microbial Functional Prediction and Distinction between Ages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Pet Product Association. Pet Industry Market Size, Trends & Ownership Statistics. 2022. Available online: https://www.americanpetproducts.org/press_industrytrends.asp (accessed on 30 June 2022).
- Hwang, E.K.; SK, K.f.g. Companion Animal in Korea Report. 2021. Available online: https://www.kbfg.com/kbresearch/report/reportView.do?reportId=2000160 (accessed on 20 June 2022).
- Ji, I.; Kim, H.; Kim, W.; Seo, G. Development strategies for the companion animal industry. Naju Korea Rural. Econ. Inst. 2017, 11–13. [Google Scholar]
- Jung, J.H. Other Livestock Statistics Survey; Ministry of Agriculture, Food and Rural Affairs: Sejong-si, Korea, 2020; p. 33.
- Wu, H.-J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Buddington, R.K.; Elnif, J.; Malo, C.; Donahoo, J.B. Activities of gastric, pancreatic, and intestinal brush-border membrane enzymes during postnatal development of dogs. Am. J. Vet. Res. 2003, 64, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The gut-brain axis: How microbiota and host inflammasome influence brain physiology and pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef]
- Ihekweazu, F.D.; Versalovic, J. Development of the pediatric gut microbiome: Impact on health and disease. Am. J. Med. Sci. 2018, 356, 413–423. [Google Scholar] [CrossRef]
- Ronan, V.; Yeasin, R.; Claud, E.C. Childhood development and the microbiome—the intestinal microbiota in maintenance of health and development of disease during childhood development. Gastroenterology 2021, 160, 495–506. [Google Scholar] [CrossRef]
- Scanes, C.G. Animal Growth. AccessSci. McGraw-Hill Educ. 2021. [Google Scholar] [CrossRef]
- Pell, J. Immunological approaches to modify growth. In Low-Fat Meats: Design Strategies and Human Implications; Academic Press: Cambridge, MA, USA, 2012; p. 303. [Google Scholar]
- Tiihonen, K.; Ouwehand, A.C.; Rautonen, N. Human intestinal microbiota and healthy ageing. Ageing Res. Rev. 2010, 9, 107–116. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Kalliomäki, M.; Carmen Collado, M.; Salminen, S.; Isolauri, E. Early differences in fecal microbiota composition in children may predict overweight. Am. J. Clin. Nutr. 2008, 87, 534–538. [Google Scholar] [CrossRef]
- Kozyrskyj, A.L.; Ernst, P.; Becker, A.B. Increased risk of childhood asthma from antibiotic use in early life. Chest 2007, 131, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Hviid, A.; Svanström, H.; Frisch, M. Antibiotic use and inflammatory bowel diseases in childhood. Gut 2011, 60, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, K.; Uchiyama, J.; Igarashi, H.; Murakami, H.; Osumi, T.; Shima, A.; Ishiahra, G.; Nasukawa, T.; Une, Y.; Sakaguchi, M. Age-related analysis of the gut microbiome in a purebred dog colony. FEMS Microbiol. Lett. 2019, 366, fnz095. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, E.; Bel Rhali, S.; Sándor, S.; Szabó, A.; Felföldi, T. Gut microbiome composition is associated with age and memory performance in pet dogs. Animals 2020, 10, 1488. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R.; Kolde, M.R.; Package ‘Pheatmap’. R Package. 2019. Available online: https://cran.r-project.org/web/packages/pheatmap/ (accessed on 18 March 2022).
- Sandri, M.; Dal Monego, S.; Conte, G.; Sgorlon, S.; Stefanon, B. Raw meat based diet influences faecal microbiome and end products of fermentation in healthy dogs. BMC Vet. Res. 2016, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Pilla, R.; Suchodolski, J.S. The role of the canine gut microbiome and metabolome in health and gastrointestinal disease. Front. Vet. Sci. 2020, 6, 498. [Google Scholar] [CrossRef] [PubMed]
- You, I.; Kim, M.J. Comparison of gut microbiota of 96 healthy dogs by individual traits: Breed, age, and body condition score. Animals 2021, 11, 2432. [Google Scholar] [CrossRef] [PubMed]
- Guard, B.C.; Mila, H.; Steiner, J.M.; Mariani, C.; Suchodolski, J.S.; Chastant-Maillard, S. Characterization of the fecal microbiome during neonatal and early pediatric development in puppies. PLoS ONE 2017, 12, e0175718. [Google Scholar]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The microbiome in early life: Implications for health outcomes. Nat. Med. 2016, 22, 713–722. [Google Scholar] [CrossRef]
- Robertson, R.C.; Manges, A.R.; Finlay, B.B.; Prendergast, A.J. The human microbiome and child growth–first 1000 days and beyond. Trends Microbiol. 2019, 27, 131–147. [Google Scholar] [CrossRef]
- Panasevich, M.R.; Kerr, K.R.; Dilger, R.N.; Fahey, G.C.; Guérin-Deremaux, L.; Lynch, G.L.; Wils, D.; Suchodolski, J.S.; Steer, J.M.; Dowd, S.E. Modulation of the faecal microbiome of healthy adult dogs by inclusion of potato fibre in the diet. Br. J. Nutr. 2015, 113, 125–133. [Google Scholar] [CrossRef]
- Lim, M.Y.; Song, E.-J.; Kang, K.S.; Nam, Y.-D. Age-related compositional and functional changes in micro-pig gut microbiome. Geroscience 2019, 41, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Masuoka, H.; Shimada, K.; Kiyosue-Yasuda, T.; Kiyosue, M.; Oishi, Y.; Kimura, S.; Yamada, A.; Hirayama, K. Transition of the intestinal microbiota of dogs with age. Biosci. Microbiota Food Health 2017, 36, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Łubiech, K.; Twarużek, M. Lactobacillus bacteria in breast milk. Nutrients 2020, 12, 3783. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lauber, C.L.; Czarnecki-Maulden, G.; Pan, Y.; Hannah, S.S. Effects of the dietary protein and carbohydrate ratio on gut microbiomes in dogs of different body conditions. MBio 2017, 8, e01703–e01716. [Google Scholar] [CrossRef]
- Bolstad, A.; Jensen, H.B.; Bakken, V. Taxonomy, biology, and periodontal aspects of Fusobacterium nucleatum. Clin. Microbiol. Rev. 1996, 9, 55–71. [Google Scholar] [CrossRef]
- Abed, J.; Maalouf, N.; Manson, A.L.; Earl, A.M.; Parhi, L.; Emgård, J.E.; Klutstein, M.; Tayeb, S.; Almogy, G.; Atlan, K.A. Colon cancer-associated Fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front. Cell. Infect. Microbiol. 2020, 10, 400. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Bui, T.P.N.; Troise, A.D.; Nijsse, B.; Roviello, G.N.; Fogliano, V.; de Vos, W.M. Intestinimonas-like bacteria are important butyrate producers that utilize Nε-fructosyllysine and lysine in formula-fed infants and adults. J. Funct. Foods 2020, 70, 103974. [Google Scholar] [CrossRef]
- Romaní-Pérez, M.; López-Almela, I.; Bullich-Vilarrubias, C.; Rueda-Ruzafa, L.; Gómez Del Pulgar, E.M.; Benítez-Páez, A.; Liebisch, G.; Lamas, J.A.; Sanz, Y. Holdemanella biformis improves glucose tolerance and regulates GLP-1 signaling in obese mice. FASEB J. 2021, 35, e21734. [Google Scholar] [CrossRef]
- Ogawa, J.; Kishino, S.; Ando, A.; Sugimoto, S.; Mihara, K.; Shimizu, S. Production of conjugated fatty acids by lactic acid bacteria. J. Biosci. Bioeng. 2005, 100, 355–364. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zheng, J.; Lin, A.; Xia, H.; Zhang, Z.; Gao, Q.; Lv, W.; Liu, H. A review: Roles of carbohydrates in human diseases through regulation of imbalanced intestinal microbiota. J. Funct. Foods 2020, 74, 104197. [Google Scholar] [CrossRef]
- Bisgaard, H.; Li, N.; Bonnelykke, K.; Chawes, B.L.K.; Skov, T.; Paludan-Müller, G.; Stokholm, J.; Smith, B.; Krogfelt, K.A. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J. Allergy Clin. Immunol. 2011, 128, 646–652.e5. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Nakayama, J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol. Int. 2017, 66, 515–522. [Google Scholar] [CrossRef]
- Reddy, K.E.; Kim, H.-R.; Jeong, J.Y.; So, K.-M.; Lee, S.; Ji, S.Y.; Kim, M.; Lee, H.-J.; Lee, S.; Kim, K.-H. Impact of breed on the fecal microbiome of dogs under the same dietary condition. J. Microbiol. Biotechnol. 2019, 29, 1947–1956. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Age (Months) | Input Reads (Average) | Total Input Reads | Output Reads (Average) | Total Output Reads | Percentage of Input Non-Chimeric (Average) |
|---|---|---|---|---|---|
| 4 | 93,915.3 | 563,492 | 52,662.83 | 315,977 | 56.03 |
| 16 | 87,963.5 | 527,781 | 49,767.17 | 298,603 | 56.55 |
| Total | 90,939.42 | 1,091,273 | 51,215 | 614,580 | 56.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.-Y.; Choi, B.-H.; Cha, J.-H.; Lim, Y.-J.; Sheet, S.; Song, M.-J.; Ko, M.-J.; Kim, N.-Y.; Kim, J.-S.; Lee, S.-J.; et al. Insight into the Fecal Microbiota Signature Associated with Growth Specificity in Korean Jindo Dogs Using 16S rRNA Sequencing. Animals 2022, 12, 2499. https://doi.org/10.3390/ani12192499
Choi S-Y, Choi B-H, Cha J-H, Lim Y-J, Sheet S, Song M-J, Ko M-J, Kim N-Y, Kim J-S, Lee S-J, et al. Insight into the Fecal Microbiota Signature Associated with Growth Specificity in Korean Jindo Dogs Using 16S rRNA Sequencing. Animals. 2022; 12(19):2499. https://doi.org/10.3390/ani12192499
Chicago/Turabian StyleChoi, So-Young, Bong-Hwan Choi, Ji-Hye Cha, Yeong-Jo Lim, Sunirmal Sheet, Min-Ji Song, Min-Jeong Ko, Na-Yeon Kim, Jong-Seok Kim, Seung-Jin Lee, and et al. 2022. "Insight into the Fecal Microbiota Signature Associated with Growth Specificity in Korean Jindo Dogs Using 16S rRNA Sequencing" Animals 12, no. 19: 2499. https://doi.org/10.3390/ani12192499
APA StyleChoi, S.-Y., Choi, B.-H., Cha, J.-H., Lim, Y.-J., Sheet, S., Song, M.-J., Ko, M.-J., Kim, N.-Y., Kim, J.-S., Lee, S.-J., Oh, S.-I., & Park, W.-C. (2022). Insight into the Fecal Microbiota Signature Associated with Growth Specificity in Korean Jindo Dogs Using 16S rRNA Sequencing. Animals, 12(19), 2499. https://doi.org/10.3390/ani12192499