
Haplotype of Wild Korean Boars Infected by Classical Swine Fever Virus Subgenotype 2.1d

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. RT-PCR Amplification of CSFV
2.2. Phylogenetic Tree and Bayesian Skyline Analysis of CSFV
2.3. PCR of the D-Loop Region of Wild Boar mtDNA
2.4. Phylogenetic and Network Analysis of Haplotypes
3. Results
3.1. The E2 Gene of CSFV from Wild Korean Boars
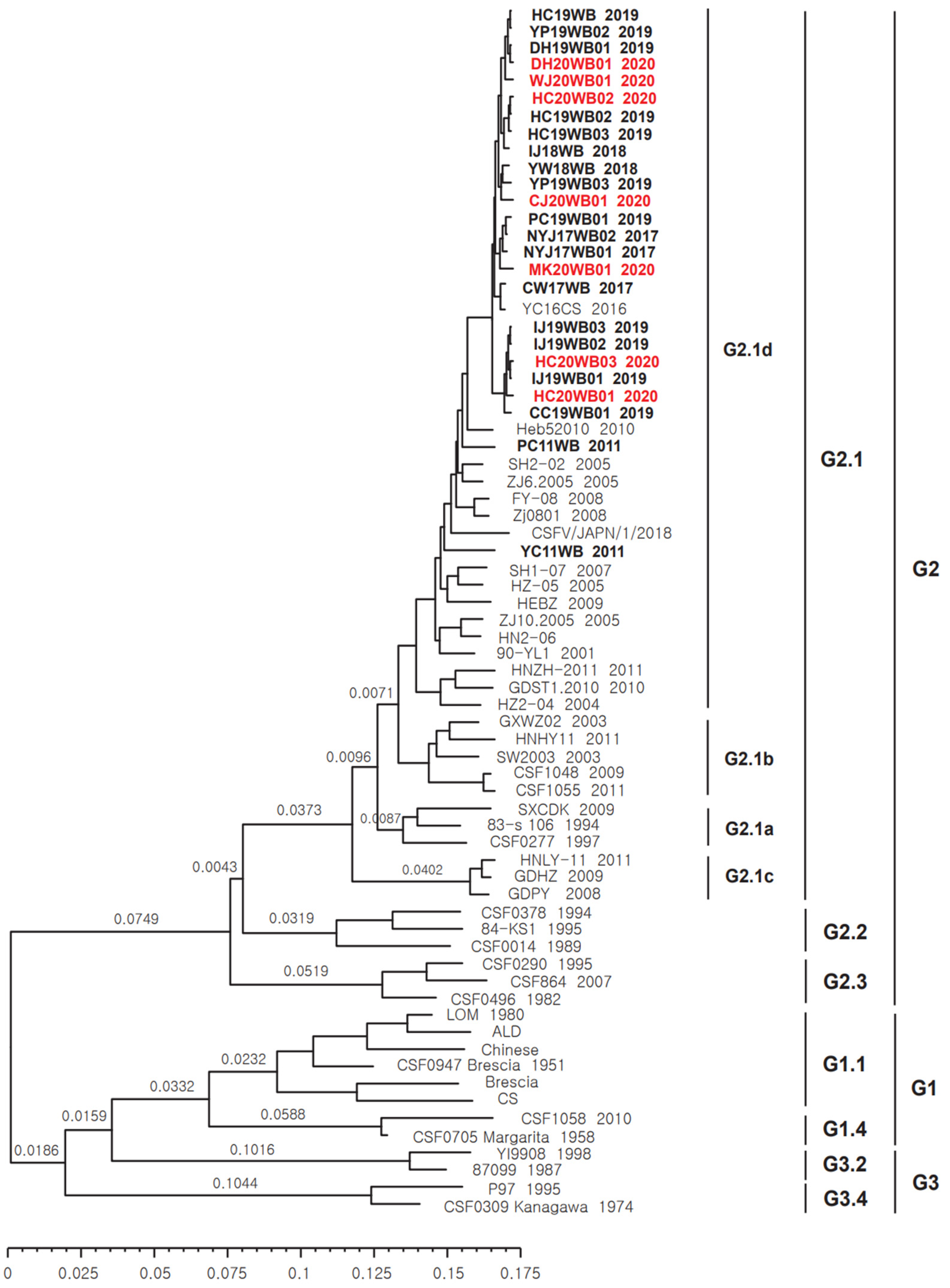
3.2. Phylogenetic Tree of CSFV
3.3. Prediction of CSF Occurrence in Wild Boars
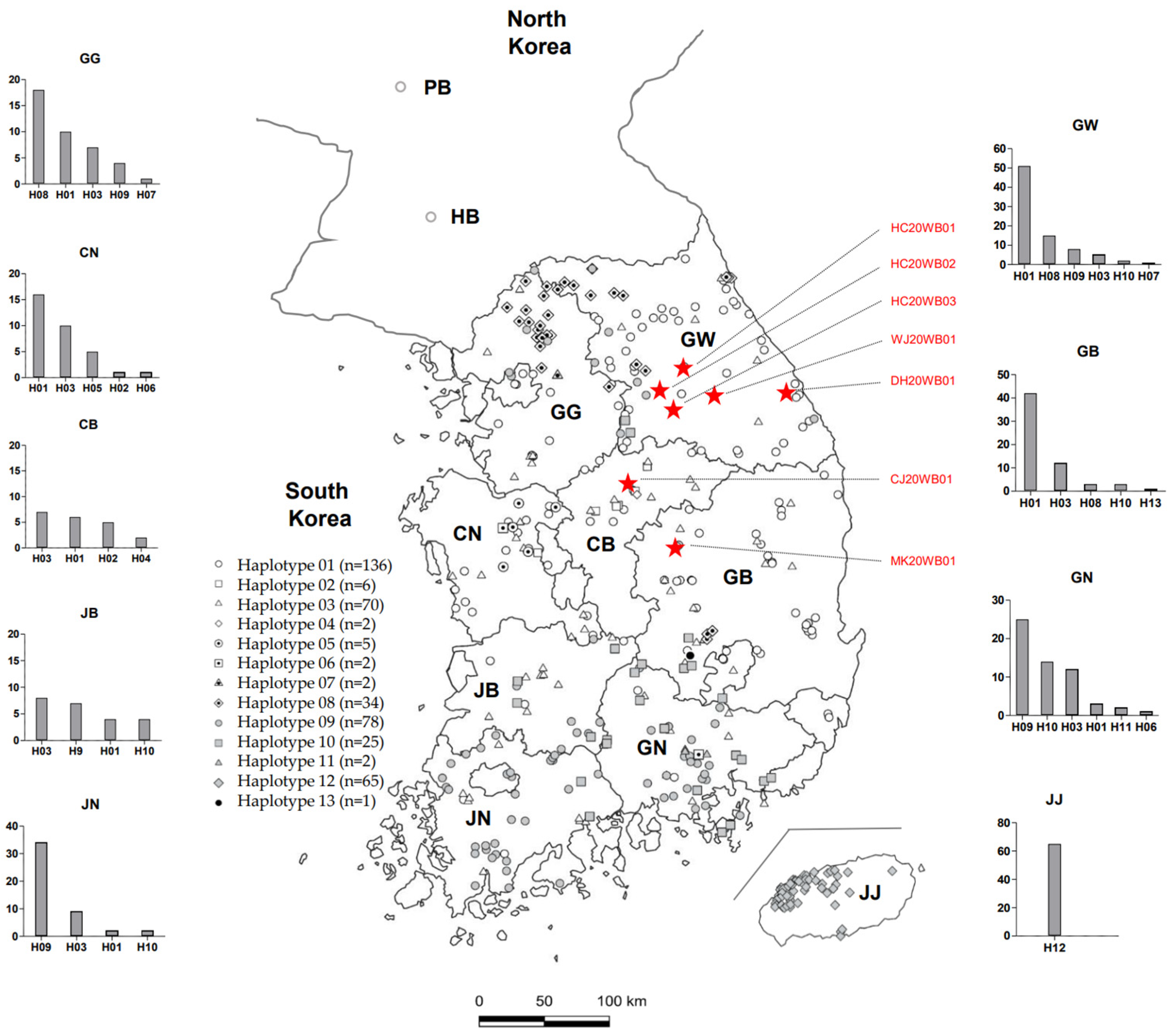
3.4. Distribution and Nucleotide Diversity of mtDNA Haplotypes among Wild Korean Boars
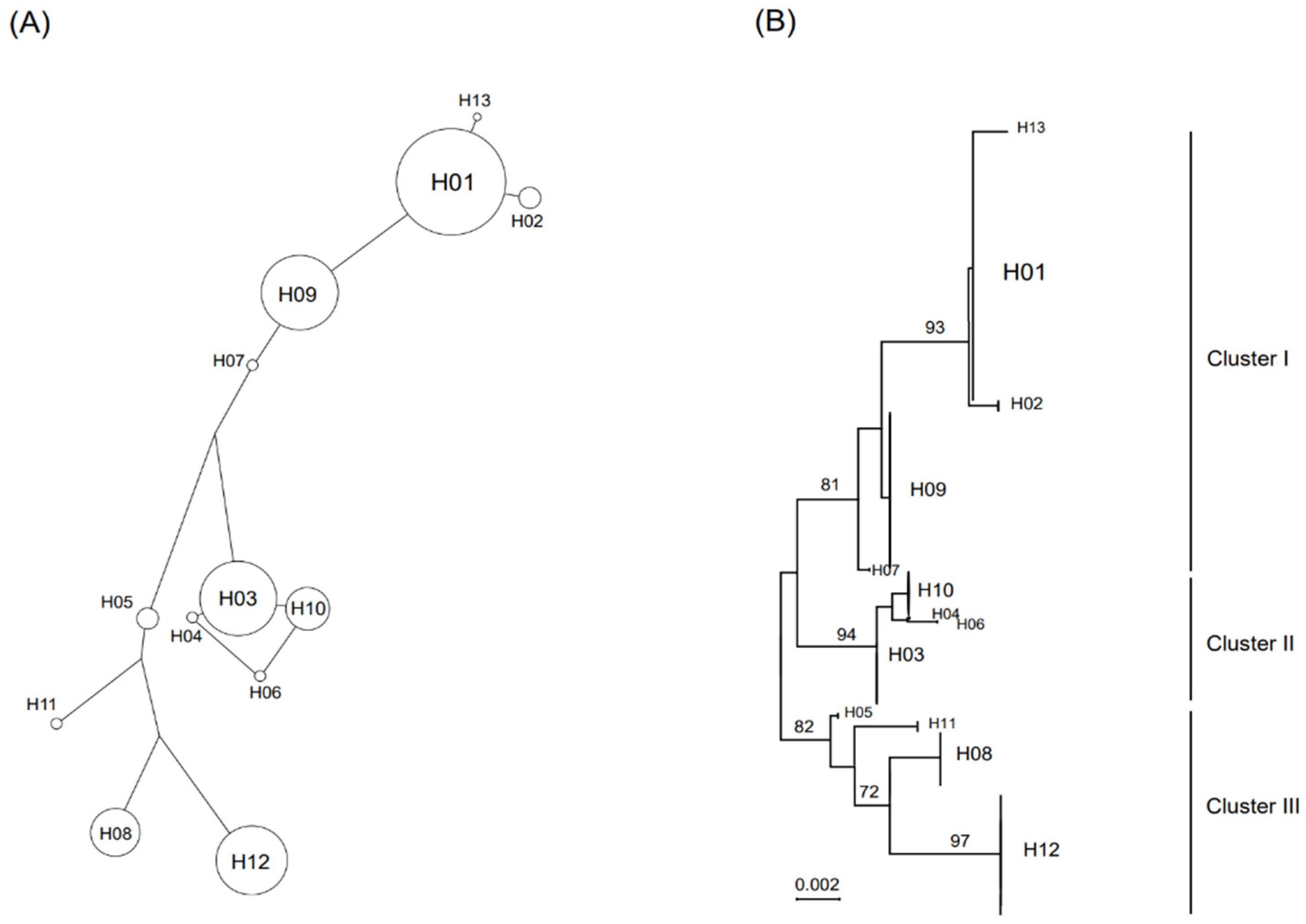
3.5. Parsimonious Median-Joining Network and Phylogenetic Analyses of mtDNA from Wild Korean Boars
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Becher, P.; Avalos Ramirez, R.; Orlich, M.; Cedillo Rosales, S.; Konig, M.; Schweizer, M.; Stalder, H.; Schirrmeier, H.; Thiel, H.J. Genetic and antigenic characterization of novel pestivirus genotypes: Implications for classification. Virology 2003, 311, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Lim, S.I.; Kim, J.J.; Cho, Y.Y.; Song, J.Y.; Cho, I.S.; Hyun, B.H.; Choi, S.H.; Kim, S.H.; Park, E.H.; et al. Surveillance of classical swine fever in wild boar in South Korea from 2010–2014. J. Vet. Med. Sci. 2016, 77, 1667–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, D.J.; Lim, S.I.; Choe, S.; Kim, K.S.; Cha, R.M.; Cho, I.S.; Song, J.Y.; Hyun, B.H.; Park, B.K. Evolutionary dynamics of classical swine fever virus in South Korea: 1987–2017. Vet. Microbiol. 2018, 225, 79–88. [Google Scholar] [CrossRef]
- Choe, S.; Cha, R.M.; Yu, D.S.; Kim, K.S.; Song, S.; Choi, S.H.; Jung, B.I.; Lim, S.I.; Hyun, B.H.; Park, B.K.; et al. Rapid spread of classical swine fever virus among South Korean wild boars in areas near the border with North Korea. Pathogens 2020, 9, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postel, A.; Moennig, V.; Becher, P. Classical swine fever in Europe--the current situation. Berl. Munch. Tierarztl. Wochenschr. 2013, 126, 468–475. [Google Scholar]
- Staubach, C.; Höreth-Böntgen, D.; Blome, S.; Fröhlich, A.; Blicke, J.; Jahn, B.; Teuffert, J.; Kramers, M. Descriptive summary of the classical swine fever control in wild boar in Germany since 2005. Berl. Munch. Tierarztl. Wochenschr. 2013, 126, 491–499. [Google Scholar]
- Zanardi, G.; Macchi, C.; Sacchi, C.; Rutili, D. Classical swine fever in wild boar in the Lombardy region of Italy from 1997 to 2002. Vet. Rec. 2003, 152, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Kameyama, K.I.; Nishi, T.; Yamada, M.; Masujin, K.; Morioka, K.; Kokuho, T.; Fukai, K. Experimental infection of pigs with a classical swine fever virus isolated in Japan for the first time in 26 years. J. Vet. Med. Sci. 2019, 81, 1277–1284. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.C.; Han, S.H.; Fang, M.; Lee, S.S.; Ko, M.S.; Lee, H.; Lim, H.T.; Yoo, C.K.; Lee, J.H.; Jeon, J.T. The robust phylogeny of Korean wild boar (Sus scrofa coreanus) using partial D-loop sequence of mtDNA. Mol. Cells 2009, 28, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Kim, J.H.; Kim, K.S.; Song, S.; Cha, R.M.; Kang, W.C.; Kim, H.J.; Park, G.N.; Shin, J.; Jo, H.N.; et al. Adverse Effects of Classical Swine Fever Virus LOM Vaccine and Jeju LOM Strains in Pregnant Sows and Specific Pathogen-Free Pigs. Pathogens 2019, 23, 9. [Google Scholar] [CrossRef] [Green Version]
- Paton, D.J.; McGoldrick, A.; Greiser-Wilke, I.; Parchariyanon, S.; Song, J.Y.; Liou, P.P.; Stadejek, T.; Lowings, J.P.; Björklund, H.; Belák, S. Genetic typing of classical swine fever virus. Vet. Microbiol. 2000, 73, 137–157. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, J. Tracer 1.5, MCMC Trace File Analyser. 2009. Available online: http://tree.bio.ed.ac.uk/software/tracer (accessed on 26 August 2021).
- Rambaut, A.; Drummond, A. TreeAnnotator v1.7.0. 2013. Available online: http://beast.bio.ed.ac.uk/treeannotator (accessed on 26 August 2021).
- Rambaut, A. FigFree. Tree Figure Drawing Tool, v.1.4.0. 2012. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 26 August 2021).
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Tajima, F. The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics 1996, 143, 1457–1465. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Harpending, H.C. Signature of Ancient Population Growth in a Low-Resolution Mitochondrial DNA Mismatch Distribution Author (s): H. C. HARPENDING Published by: Wayne State University Press. Hum. Biol. 2013, 66, 591–600. [Google Scholar]
- Bandelt, H.J.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Moennig, V. The control of classical swine fever in wild boar. Front. Microbiol. 2015, 6, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, S.; Staubach, C.; Blome, S.; Guberti, V.; Thulke, H.H.; Vos, A.; Koenen, F.; Le Potier, M.F. Controlling of CSFV in European wild boar using oral vaccination: A review. Front. Microbiol. 2015, 6, 1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritzemeier, J.; Teuffert, J.; Greiser-Wilke, I.; Staubach, C.; Schluter, H.; Moennig, V. Epidemiology of classical swine fever in Germany in the 1990s. Vet. Microbiol. 2000, 77, 29–41. [Google Scholar] [CrossRef]
- Ito, S.; Jurado, C.; Bosch, J.; Ito, M.; Sanchez-Vizcaino, J.M.; Isoda, N.; Sakoda, A.Y. Role of Wild Boar in the Spread of Classical Swine Fever in Japan. Pathogens 2019, 8, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Hayama, Y.; Murato, Y.; Sawai, K.; Yamaguchi, E.; Yamamoto, T. Epidemiological analysis of classical swine fever in wild boars in Japan. BMC Vet. Res. 2021, 17, 188. [Google Scholar] [CrossRef]
- Hayama, Y.; Sawai, K.; Yoshinori, M.; Yamaguchi, E.; Shimizu, Y.; Yamamoto, T. Pig farm vaccination against classical swine fever reduces the risk of transmission from wild boar. Prev. Vet. Med. 2022, 198, 105554. [Google Scholar] [CrossRef]
- Jo, Y.S.; Gortázar, C. African Swine Fever in wild boar: Assessing interventions in South Korea. Transbound. Emerg. Dis. 2021, 68, 2878–2889. [Google Scholar] [CrossRef] [PubMed]
- Postel, A.; Nishi, T.; Kameyama, K.I.; Meyer, D.; Suckstorff, O.; Fukai, K.; Becher, P. Reemergence of classical swine fever, Japan, 2018. Emerg. Infect. Dis. 2019, 25, 1228–1231. [Google Scholar] [CrossRef] [Green Version]
- Nishi, T.; Kameyama, K.I.; Kato, T.; Fukai, K. Genome sequence of a classical swine fever virus of subgenotype 2.1, isolated from a pig in Japan in 2018. Microbiol. Resour. Announc. 2019, 8, e01362-18. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.I.; Kim, Y.K.; Lim, J.; Han, S.H.; Hyun, H.S.; Kim, K.S.; Hyun, B.H.; Kim, J.J.; Cho, I.S.; Song, J.Y.; et al. Antigenic characterization of classical swine fever virus YC11WB isolates from wild boar. J. Vet. Sci. 2017, 18, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, N.; Nishimura, M. Genetic profile and serosurvey for virus infections of Japanese wild boars in Shikoku Island. J. Vet. Med. Sci. 2005, 67, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanobe, T.; Okumura, N.; Ishiguro, N.; Nakano, M.; Matsui, A.; Sahara, M.; Komatsu, M. Genetic relationship and distribution of the Japanese wild boar (Sus scrofa leucomystax) and Ryukyu wild boar (Sus scrofa riukiuanus) analysed by mitochondrial DNA. Mol. Ecol. 1999, 8, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Watanobe, T.; Ishiguro, N.; Nakano, M.; Matsui, A.; Hongo, H.; Yamazaki, K.; Takahashi, O. Prehistoric Sado Island populations of Sus scrofa distinguished from contemporary Japanese wild boar by ancient mitochondrial DNA. Zoolog. Sci. 2004, 21, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Okumura, N.; Ishiguro, N.; Nakano, M.; Hirai, K.; Matsui, A.; Sahara, M. Geographic population structure and sequence divergence in the mitochondrial DNA control region of the Japanese wild boar (Sus scrofa leucomystax), with reference to those of domestic pigs. Biochem. Genet. 1996, 34, 179–189. [Google Scholar] [CrossRef]
- Charoensook, R.; Brening, B.; Gatphayak, K.; Knorr, C. Further resolution of porcine phylogeny in Southeast Asia by Thai mtDNA haplotypes. Anim. Genet. 2011, 42, 445–450. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total | North Korea | South Korea | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PB a | HB | GG | GW | CN | CB | GB | GN | JB | JN | JJ | ||
| CSFV | 7/2140 b | - | - | 0/141 | 5/201 | 0/236 | 1/267 | 1/486 | 0/363 | 0/320 | 0/82 | 0/44 |
| Haplotype | 12/347 c | - | - | 5/30 | 6/71 | 5/32 | 4/20 | 3/54 | 6/56 | 4/23 | 4/35 | 1/26 |
| 7/81 d | 1/1 | 1/1 | 4/8 | 3/11 | 1/1 | - | 3/7 | 1/1 | - | 1/12 | 1/39 | |
| 13/428 e | 1/1 | 1/1 | 5/38 | 6/82 | 5/33 | 4/20 | 5/61 | 6/57 | 4/23 | 4/47 | 1/65 | |
| 2011 Strain | Isolation Year (Number of Strains) | |||
|---|---|---|---|---|
| 2017 (3) a | 2018 (2) b | 2019 (11) c | 2020 (7) d | |
| YC11WB | 95.95 ± 0.35 e 98.0 ± 0.4 f | 95.6 ± 0.3 97.85 ± 0.55 | 95.9 ± 0.2 98.15 ± 0.55 | 95.95 ± 0.15 98.4 ± 0.3 |
| PC11WB | 97.35 ± 0.25 98.8 ± 0.4 | 96.75 ± 0.25 98.4 ± 0.3 | 97.15 ± 0.15 98.95 ± 0.55 | 97.2 ± 0.1 99.2 ± 0.3 |
| Country | Location | No. of Wild Boars | mtDNA Control Region a | ||||
|---|---|---|---|---|---|---|---|
| Hd | Π (%) | Tajima’s D | Fu’s Fs | r | |||
| Korea b | 11 c | 428 d | 0.807 | 0.00925 | 2.63354 | 8.450 | 0.0809 |
| South Korea | GG e | 38 | 0.727 | 0.00932 | 2.89214 | 8.371 | 0.1799 |
| GW | 82 | 0.573 | 0.00673 | 1.48833 | 7.044 | 0.3038 | |
| CN | 33 | 0.669 | 0.00750 | 1.93526 | 6.018 | 0.2823 | |
| CB | 20 | 0.753 | 0.00758 | 2.91511 | 5.791 | 0.1907 | |
| GB | 61 | 0.490 | 0.00620 | 0.70848 | 6.840 | 0.3498 | |
| GN | 57 | 0.711 | 0.00605 | 0.80308 | 4.975 | 0.1404 | |
| JB | 23 | 0.759 | 0.00635 | 2.01929 | 5.320 | 0.1511 | |
| JN | 47 | 0.446 | 0.00371 | 0.34085 | 4.296 | 0.4301 | |
| JJ | 65 | 0.030 | 0.00054 | 0.00713 | 1.422 | 0.9421 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choe, S.; Kim, K.-S.; Park, G.-N.; Song, S.; Shin, J.; Hyun, B.-H.; An, D.-J. Haplotype of Wild Korean Boars Infected by Classical Swine Fever Virus Subgenotype 2.1d. Animals 2022, 12, 2670. https://doi.org/10.3390/ani12192670
Choe S, Kim K-S, Park G-N, Song S, Shin J, Hyun B-H, An D-J. Haplotype of Wild Korean Boars Infected by Classical Swine Fever Virus Subgenotype 2.1d. Animals. 2022; 12(19):2670. https://doi.org/10.3390/ani12192670
Chicago/Turabian StyleChoe, SeEun, Ki-Sun Kim, Gyu-Nam Park, Sok Song, Jihye Shin, Bang-Hun Hyun, and Dong-Jun An. 2022. "Haplotype of Wild Korean Boars Infected by Classical Swine Fever Virus Subgenotype 2.1d" Animals 12, no. 19: 2670. https://doi.org/10.3390/ani12192670