
Expression Profile of Housekeeping Genes and Tissue-Specific Genes in Multiple Tissues of Pigs
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Samples
2.2. Data Quality Control, Alignment, and Processing
2.3. Gene Expression Pattern among Tissues
2.4. Detection of Housing-Keeping Genes
2.5. Detection of Tissue-Specifc Gene
2.6. Co-Expression Network Analysis
2.7. Functional Enrichment and Protein–Protein Interaction Analysis and the Validation of HKGs and TSGs
3. Results
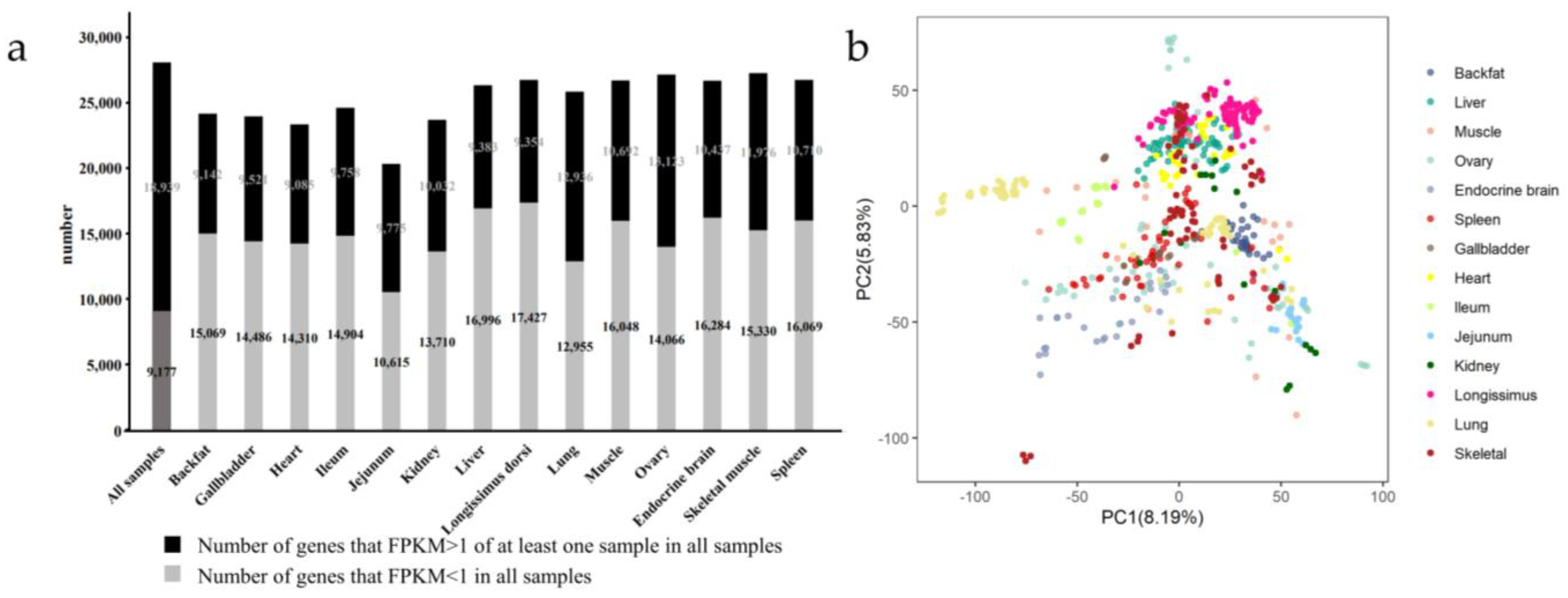
3.1. Detection of Genes in Multiple Tissues
3.2. The Profiles of Housekeeping Gene across Pig Tissues
3.3. Tissue-Specific Genes Expression Patterns across Pig Tissues
3.4. Co-Expression Gene Network across Pigs Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Monguio-Tortajada, M.; Prat-Vidal, C.; Martinez-Falguera, D.; Teis, A.; Soler-Botija, C.; Courageux, Y.; Munizaga-Larroude, M.; Moron-Font, M.; Bayes-Genis, A.; Borras, F.E.; et al. Acellular cardiac scaffolds enriched with MSC-derived extracellular vesicles limit ventricular remodelling and exert local and systemic immunomodulation in a myocardial infarction porcine model. Theranostics 2022, 12, 4656–4670. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schurmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neff, E.P. Cancer modeling thinks big with the pig. Lab. Anim. 2019, 48, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Holm, I.E.; Alstrup, A.K.; Luo, Y. Genetically modified pig models for neurodegenerative disorders. J. Pathol. 2016, 238, 267–287. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Kern, C.; Wang, Y.; Xu, X.; Pan, Z.; Halstead, M.; Chanthavixay, G.; Saelao, P.; Waters, S.; Xiang, R.; Chamberlain, A.; et al. Functional annotations of three domestic animal genomes provide vital resources for comparative and agricultural research. Nat. Commun. 2021, 12, 1821. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Hu, H.; Wang, H.; Wang, C.; Lin, H.; Zhao, X. Dynamic transcriptome profiles of postnatal porcine skeletal muscle growth and development. BMC Genom. Data 2021, 22, 32. [Google Scholar] [CrossRef]
- Albuquerque, A.; Ovilo, C.; Nunez, Y.; Benitez, R.; Lopez-Garcia, A.; Garcia, F.; Felix, M.; Laranjo, M.; Charneca, R.; Martins, J.M. Comparative Transcriptomic Analysis of Subcutaneous Adipose Tissue from Local Pig Breeds. Genes 2020, 11, 422. [Google Scholar] [CrossRef] [Green Version]
- Bertho, N.; Meurens, F. The pig as a medical model for acquired respiratory diseases and dysfunctions: An immunological perspective. Mol. Immunol. 2021, 135, 254–267. [Google Scholar] [CrossRef]
- Lim, K.S.; Cheng, J.; Putz, A.; Dong, Q.; Bai, X.; Beiki, H.; Tuggle, C.K.; Dyck, M.K.; Canada, P.G.; Fortin, F.; et al. Quantitative analysis of the blood transcriptome of young healthy pigs and its relationship with subsequent disease resilience. BMC Genom. 2021, 22, 614. [Google Scholar] [CrossRef]
- Gonzalez-Prendes, R.; Quintanilla, R.; Marmol-Sanchez, E.; Pena, R.N.; Ballester, M.; Cardoso, T.F.; Manunza, A.; Casellas, J.; Canovas, A.; Diaz, I.; et al. Comparing the mRNA expression profile and the genetic determinism of intramuscular fat traits in the porcine gluteus medius and longissimus dorsi muscles. BMC Genom. 2019, 20, 170. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, Y.; Canela-Xandri, O.; Wang, S.; Yu, Y.; Cai, W.; Li, B.; Xiang, R.; Chamberlain, A.J.; Pairo-Castineira, E.; et al. A multi-tissue atlas of regulatory variants in cattle. Nat. Genet. 2022, 54, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.L.; Bush, S.J.; McCulloch, M.; Farquhar, I.L.; Young, R.; Lefevre, L.; Pridans, C.; Tsang, H.G.; Wu, C.; Afrasiabi, C.; et al. A high resolution atlas of gene expression in the domestic sheep (Ovis aries). PLoS Genet. 2017, 13, e1006997. [Google Scholar] [CrossRef] [Green Version]
- Kanis, E.; De Greef, K.H.; Hiemstra, A.; van Arendonk, J.A. Breeding for societally important traits in pigs. J. Anim. Sci. 2005, 83, 948–957. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Ye, S.; Chen, Z.; Pan, X.; Huang, S.; Li, Z.; Zhong, Y.; Gao, N.; Zhang, H.; Li, J.; et al. Dynamic DNA methylation of ovaries during pubertal transition in gilts. BMC Genom. 2019, 20, 510. [Google Scholar] [CrossRef] [PubMed]
- Salavati, M.; Woolley, S.A.; Cortes, A.Y.; Halstead, M.M.; Stenhouse, C.; Johnsson, M.; Ashworth, C.J.; Archibald, A.L.; Donadeu, F.X.; Hassan, M.A.; et al. Profiling of open chromatin in developing pig (Sus scrofa) muscle to identify regulatory regions. G3 2022, 12, jkab424. [Google Scholar] [CrossRef]
- Chen, S.; Liu, S.; Mi, S.; Li, W.; Zhang, S.; Ding, X.; Yu, Y. Comparative Analyses of Sperm DNA Methylomes among Three Commercial Pig Breeds Reveal Vital Hypomethylated Regions Associated with Spermatogenesis and Embryonic Development. Front. Genet. 2021, 12, 740036. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes are compact. Trends Genet. 2003, 19, 362–365. [Google Scholar] [CrossRef] [Green Version]
- Butte, A.J.; Dzau, V.J.; Glueck, S.B. Further defining housekeeping, or “maintenance,” genes focus on “A compendium of gene expression in normal human tissues”. Physiol. Genom. 2001, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Hounkpe, B.W.; Chenou, F.; de Lima, F.; De Paula, E.V. HRT Atlas v1.0 database: Redefining human and mouse housekeeping genes and candidate reference transcripts by mining massive RNA-seq datasets. Nucleic Acids Res. 2021, 49, D947–D955. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, T.; Niu, Q.; Xu, L.; Chen, Y.; Gao, X.; Gao, H.; Zhang, L.; Liu, G.E.; Li, J.; et al. Transcriptional atlas analysis from multiple tissues reveals the expression specificity patterns in beef cattle. BMC Biol. 2022, 20, 79. [Google Scholar] [CrossRef] [PubMed]
- Joshi, C.J.; Ke, W.; Drangowska-Way, A.; O’Rourke, E.J.; Lewis, N.E. What are housekeeping genes? PLoS Comput. Biol. 2022, 18, e1010295. [Google Scholar] [CrossRef] [PubMed]
- She, X.; Rohl, C.A.; Castle, J.C.; Kulkarni, A.V.; Johnson, J.M.; Chen, R. Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genom. 2009, 10, 269. [Google Scholar] [CrossRef] [Green Version]
- Sonawane, A.R.; Platig, J.; Fagny, M.; Chen, C.Y.; Paulson, J.N.; Lopes-Ramos, C.M.; DeMeo, D.L.; Quackenbush, J.; Glass, K.; Kuijjer, M.L. Understanding Tissue-Specific Gene Regulation. Cell Rep. 2017, 21, 1077–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, W.H. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol. Biol. Evol. 2004, 21, 236–239. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, e17. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, Z.; Ranjbaran, J.; Safarpour, H.; Nomiri, S.; Salmani, F.; Chamani, E.; Larki, P.; Brunetti, O.; Silvestris, N.; Tavakoli, T. Identification of early diagnostic biomarkers via WGCNA in gastric cancer. Biomed. Pharm. 2022, 145, 112477. [Google Scholar] [CrossRef]
- Sheng, H.; Pan, C.; Wang, S.; Yang, C.; Zhang, J.; Hu, C.; Hu, H.; Feng, X.; Yang, M.; Lei, Z.; et al. Weighted Gene Co-Expression Network Analysis Identifies Key Modules and Central Genes Associated with Bovine Subcutaneous Adipose Tissue. Front. Vet. Sci. 2022, 9, 914848. [Google Scholar] [CrossRef]
- Xu, C.; Wang, X.; Zhou, S.; Wu, J.; Geng, Q.; Ruan, D.; Qiu, Y.; Quan, J.; Ding, R.; Cai, G.; et al. Brain Transcriptome Analysis Reveals Potential Transcription Factors and Biological Pathways Associated with Feed Efficiency in Commercial DLY Pigs. DNA Cell Biol. 2021, 40, 272–282. [Google Scholar] [CrossRef]
- Wu, Z.; Hai, E.; Di, Z.; Ma, R.; Shang, F.; Wang, Y.; Wang, M.; Liang, L.; Rong, Y.; Pan, J.; et al. Using WGCNA (weighted gene co-expression network analysis) to identify the hub genes of skin hair follicle development in fetus stage of Inner Mongolia cashmere goat. PLoS ONE 2020, 15, e243507. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Ha, M.; Kraushaar, D.C.; Zhao, K. Genome-wide analysis of H3.3 dissociation reveals high nucleosome turnover at distal regulatory regions of embryonic stem cells. Epigenet. Chromatin 2014, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Liu, H.; Huang, L.; Zhang, X.; Dong, X.; Song, W.; Zhao, H.; Lai, J. Long-range interactions between proximal and distal regulatory regions in maize. Nat. Commun. 2019, 10, 2633. [Google Scholar] [CrossRef] [Green Version]
- de Jonge, H.J.; Fehrmann, R.S.; de Bont, E.S.; Hofstra, R.M.; Gerbens, F.; Kamps, W.A.; de Vries, E.G.; van der Zee, A.G.; Te, M.G.; ter Elst, A. Evidence based selection of housekeeping genes. PLoS ONE 2007, 2, e898. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Sun, F.; Zhao, Y.; Shan, L.; Lou, H. Identification of Susceptibility Modules and Genes for Cardiovascular Disease in Diabetic Patients Using WGCNA Analysis. J. Diabetes Res. 2020, 2020, 4178639. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; Gao, Y.; Yin, H.; Bai, Z.; Liu, S.; Zeng, H.; Bai, L.; Cai, Z.; Zhao, B.; Li, X.; et al. A compendium of genetic regulatory effects across pig tissues. bioRxiv 2022. [Google Scholar] [CrossRef]
- Herrero-Medrano, J.M.; Megens, H.J.; Groenen, M.A.; Ramis, G.; Bosse, M.; Perez-Enciso, M.; Crooijmans, R.P. Conservation genomic analysis of domestic and wild pig populations from the Iberian Peninsula. BMC Genet. 2013, 14, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Cai, W.; Liu, S.; Canela-Xandri, O.; Gao, Y.; Jiang, J.; Rawlik, K.; Li, B.; Schroeder, S.G.; Rosen, B.D.; et al. Comprehensive analyses of 723 transcriptomes enhance genetic and biological interpretations for complex traits in cattle. Genome Res. 2020, 30, 790–801. [Google Scholar] [CrossRef]
- Battle, A.; Brown, C.D.; Engelhardt, B.E.; Montgomery, S.B. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar]
- Jiang, W.; Chen, L. Tissue Specificity of Gene Expression Evolves Across Mammal Species. J. Comput. Biol. 2022, 29, 880–891. [Google Scholar] [CrossRef]
- Summers, K.M.; Bush, S.J.; Wu, C.; Su, A.I.; Muriuki, C.; Clark, E.L.; Finlayson, H.A.; Eory, L.; Waddell, L.A.; Talbot, R.; et al. Functional Annotation of the Transcriptome of the Pig, Sus scrofa, Based Upon Network Analysis of an RNAseq Transcriptional Atlas. Front. Genet. 2019, 10, 1355. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, C.; Adeola, A.C.; Sulaiman, X.; Xie, H.B.; Zhang, Y.P. Artificial selection drives differential gene expression during pig domestication. J. Genet. Genom. 2019, 46, 97–100. [Google Scholar] [CrossRef]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Sohail, M.; Chua, R.; Sim, S.K.; Nik, A.N. Jejunal gastrointestinal stromal tumour masquerading as an ovarian cancer: A case report. Med. J. Malays. 2022, 77, 125–127. [Google Scholar]
- Zheng, J.; Wang, J.; Pouliot, M.; Authier, S.; Zhou, D.; Loose, D.S.; Hauer-Jensen, M. Gene expression profiling in non-human primate jejunum, ileum and colon after total-body irradiation: A comparative study of segment-specific molecular and cellular responses. BMC Genom. 2015, 16, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Shang, T.; Huang, Y.; Wang, S.; Liu, H.; Wang, J.; Wang, Y.; Ji, H.; Zhang, R. Gene expression profile changes in the jejunum of weaned piglets after oral administration of Lactobacillus or an antibiotic. Sci. Rep. 2017, 7, 15816. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Nakagawa, M.; Herbert, J.; Lomax, M.I.; Grossman, L.I.; Sherbany, A.A.; Miranda, A.F.; DiMauro, S.; Schon, E.A. Isolation of a cDNA clone encoding subunit IV of human cytochrome c oxidase. Gene 1987, 55, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.W.; Yi, Y.J.; Sutovsky, M.; van Leeuwen, F.W.; Conant, G.; Sutovsky, P. Identification and characterization of RING-finger ubiquitin ligase UBR7 in mammalian spermatozoa. Cell Tissue Res. 2014, 356, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Gandre, S.; Bercovich, Z.; Kahana, C. Mitochondrial localization of antizyme is determined by context-dependent alternative utilization of two AUG initiation codons. Mitochondrion 2003, 2, 245–256. [Google Scholar] [CrossRef]
- Thorson, J.F.; Heidorn, N.L.; Ryu, V.; Czaja, K.; Nonneman, D.J.; Barb, C.R.; Hausman, G.J.; Rohrer, G.A.; Prezotto, L.D.; McCosh, R.B.; et al. Relationship of neuropeptide FF receptors with pubertal maturation of gilts. Biol. Reprod. 2017, 96, 617–634. [Google Scholar] [CrossRef] [Green Version]
- D’Aquino, A.E.; Kim, D.S.; Jewett, M.C. Engineered Ribosomes for Basic Science and Synthetic Biology. Annu. Rev. Chem. Biomol. Eng. 2018, 9, 311–340. [Google Scholar] [CrossRef]
- Wang, K.; Vijay, V.; Fuscoe, J.C. Stably Expressed Genes Involved in Basic Cellular Functions. PLoS ONE 2017, 12, e170813. [Google Scholar] [CrossRef] [Green Version]
- Agueda-Oyarzabal, M.; Emanuelli, B. Immune Cells in Thermogenic Adipose Depots: The Essential but Complex Relationship. Front. Endocrinol. 2022, 13, 839360. [Google Scholar] [CrossRef]
- Kadenbach, B. Introduction to mitochondrial oxidative phosphorylation. Adv. Exp. Med. Biol. 2012, 748, 1–11. [Google Scholar]
- O’Reilly, A.J.; Dacks, J.B.; Field, M.C. Evolution of the karyopherin-beta family of nucleocytoplasmic transport factors: Ancient origins and continued specialization. PLoS ONE 2011, 6, e19308. [Google Scholar]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes, revisited. Trends Genet. 2013, 29, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Adashi, E.Y. Endocrinology of the ovary. Hum. Reprod. 1994, 9, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Annie, L.; Gurusubramanian, G.; Roy, V.K. Inhibition of visfatin/NAMPT affects ovarian proliferation, apoptosis, and steroidogenesis in pre-pubertal mice ovary. J. Steroid Biochem. Mol. Biol. 2020, 204, 105763. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Walsh, M.; Bokov, A.; Ikeno, Y.; Jang, Y.C.; Perez, V.I.; Van Remmen, H.; Richardson, A. Liver specific expression of Cu/ZnSOD extends the lifespan of Sod1 null mice. Mech. Ageing Dev. 2016, 154, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Danner, S.; Lohse, M.J. Phosducin is a ubiquitous G-protein regulator. Proc. Natl. Acad. Sci. USA 1996, 93, 10145–10150. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Wu, M.; Guo, R.; Yan, K.; Lei, J.; Gao, N.; Yang, M. The architecture of the mammalian respirasome. Nature 2016, 537, 639–643. [Google Scholar] [CrossRef]
- Wang, H.; Jiao, P.; Zhang, X.; Xing, H. Quantitative proteomic analysis of trachea in fatting pig exposed to ammonia. J. Proteom. 2021, 247, 104330. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, Y.; Wu, D.; Hainan, L. The effect of heat stress on the epidermal growth factor (EGF)-mediated intracellular signaling, and changes cell behavior on swine testicular cell. Anim. Biotechnol. 2022, 1–9. [Google Scholar] [CrossRef]
- Wang, J.; Khan, S.U.; Cao, P.; Chen, X.; Wang, F.; Zou, D.; Li, H.; Zhao, H.; Xu, K.; Jiao, D.; et al. Construction of PIK3C3 Transgenic Pig and Its Pathogenesis of Liver Damage. Life 2022, 12, 630. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.; Cai, J.; Wang, Y.; Xu, D.; Jiang, Y.; Gong, W.; Tian, Y.; Shen, Q.; Zhang, Z.; Yuan, X.; et al. Expression Profile of Housekeeping Genes and Tissue-Specific Genes in Multiple Tissues of Pigs. Animals 2022, 12, 3539. https://doi.org/10.3390/ani12243539
Pan X, Cai J, Wang Y, Xu D, Jiang Y, Gong W, Tian Y, Shen Q, Zhang Z, Yuan X, et al. Expression Profile of Housekeeping Genes and Tissue-Specific Genes in Multiple Tissues of Pigs. Animals. 2022; 12(24):3539. https://doi.org/10.3390/ani12243539
Chicago/Turabian StylePan, Xiangchun, Jiali Cai, Yifei Wang, Dantong Xu, Yao Jiang, Wentao Gong, Yuhan Tian, Qingpeng Shen, Zhe Zhang, Xiaolong Yuan, and et al. 2022. "Expression Profile of Housekeeping Genes and Tissue-Specific Genes in Multiple Tissues of Pigs" Animals 12, no. 24: 3539. https://doi.org/10.3390/ani12243539
APA StylePan, X., Cai, J., Wang, Y., Xu, D., Jiang, Y., Gong, W., Tian, Y., Shen, Q., Zhang, Z., Yuan, X., & Li, J. (2022). Expression Profile of Housekeeping Genes and Tissue-Specific Genes in Multiple Tissues of Pigs. Animals, 12(24), 3539. https://doi.org/10.3390/ani12243539