
Comparison of the Intestinal Structure and Intestinal Microbiome between Two Geographically Isolated Populations of Culter alburnus

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
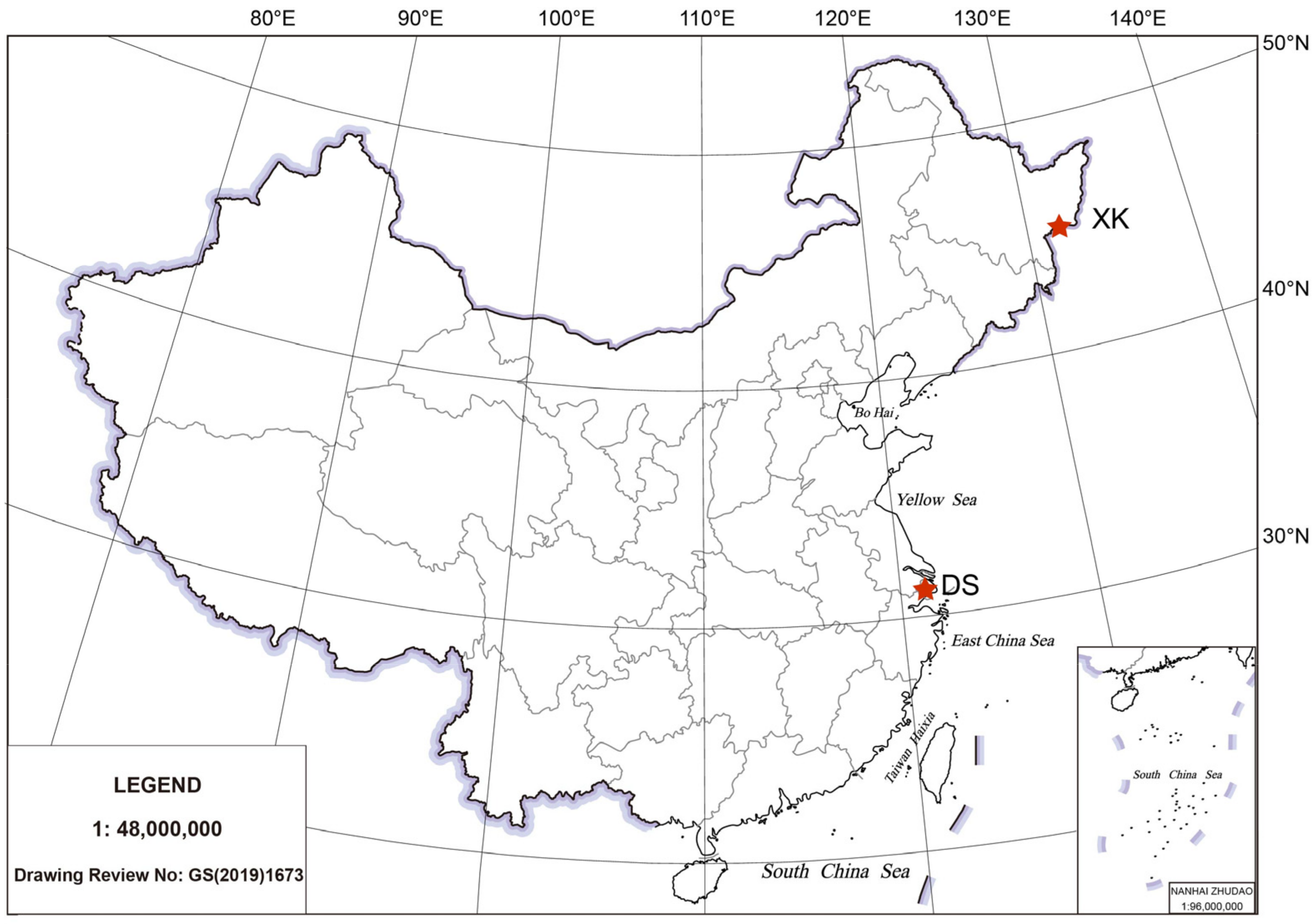
2.1. Animal Sampling
2.2. Intestinal Structure and Digestive Enzyme Activity Evaluation
2.3. Sequencing of 16s rRNA and Data Analysis
3. Results
3.1. Comparison of Intestinal Structure and Digestive Enzyme Activity
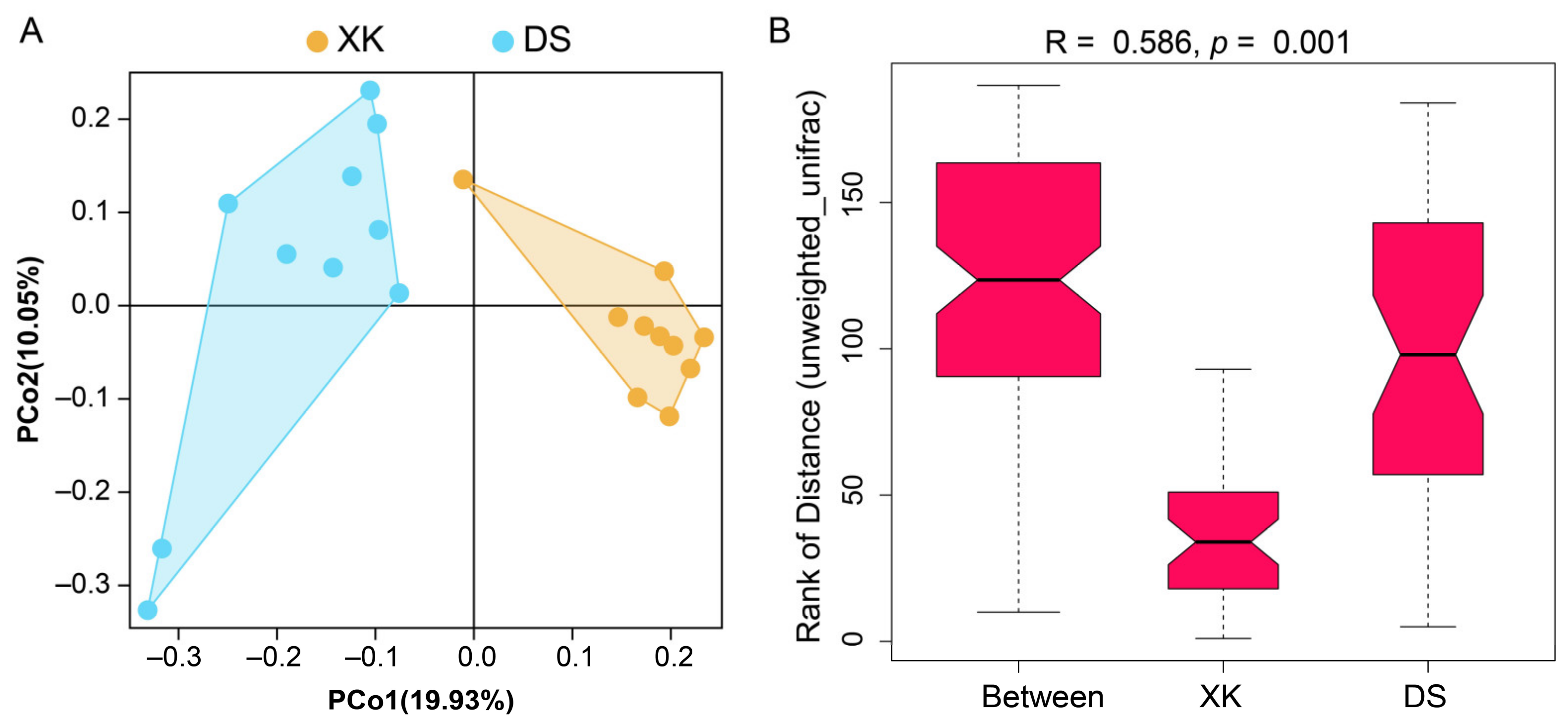
3.2. Sequencing and Diversity Estimate of Gut Microbiome between the Two Populations
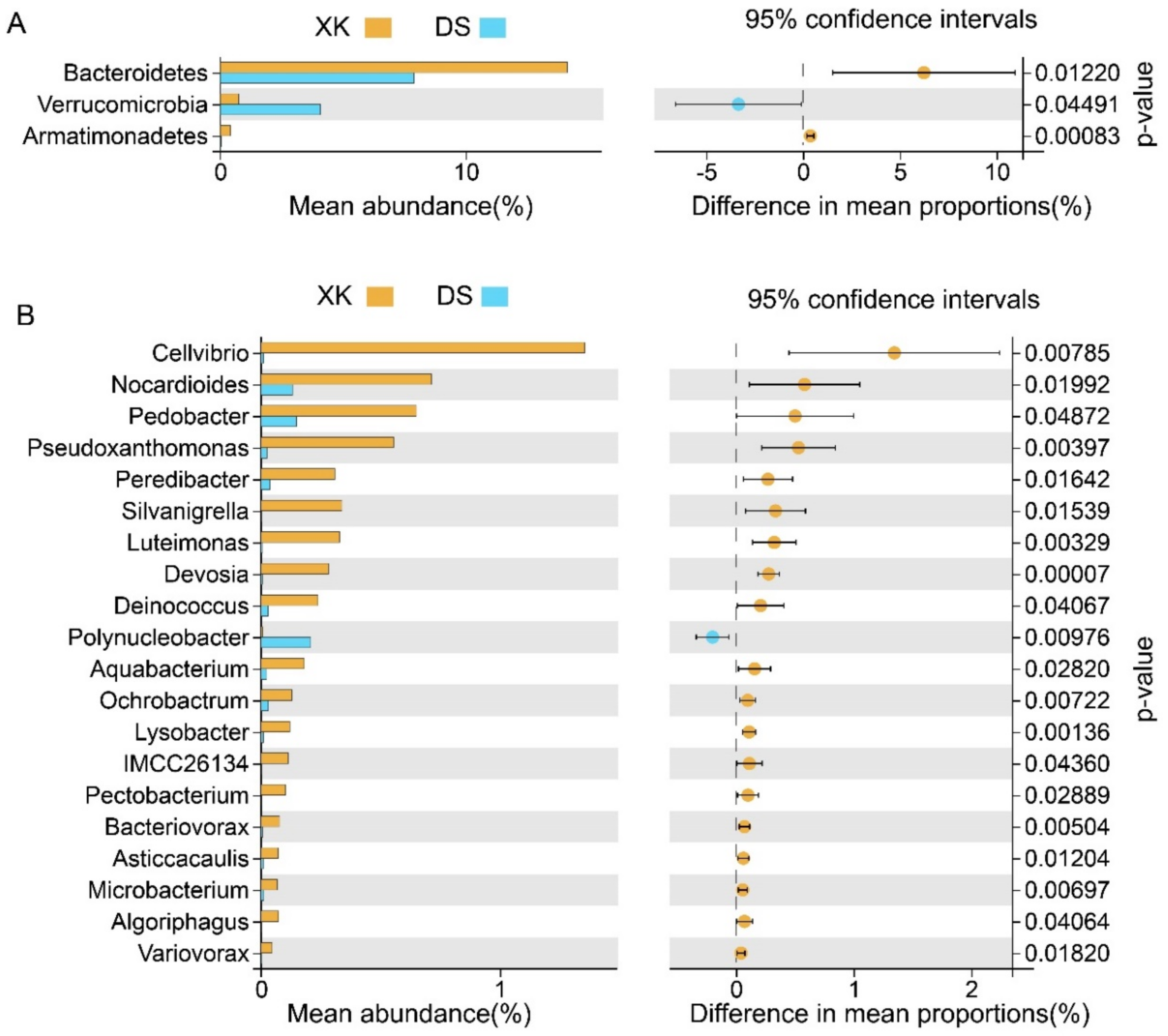
3.3. Comparison of Intestinal Microbiome Compositions between the Two Populations
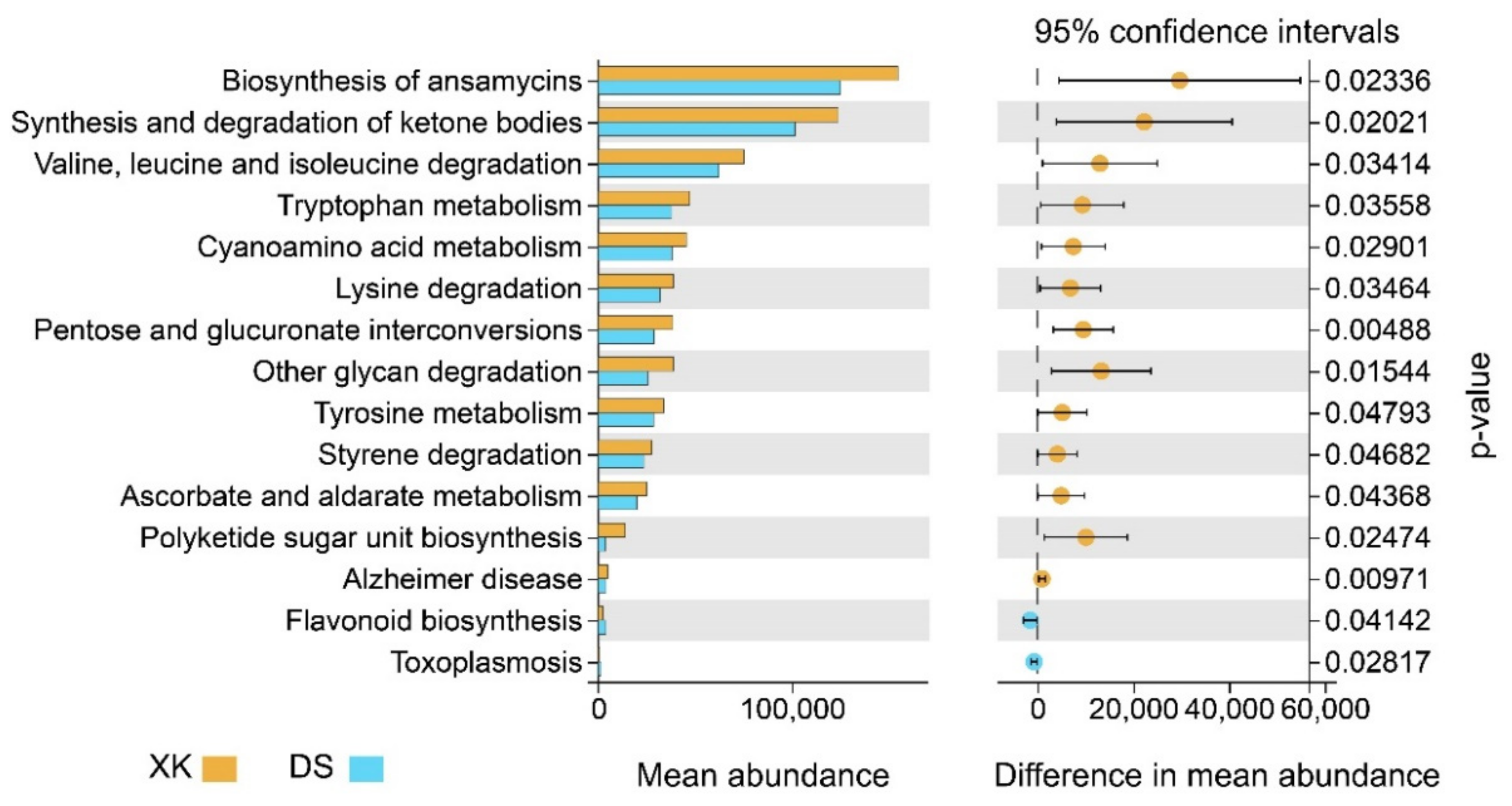
3.4. Functional Divergence between the Two Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hendry, A.P.; Nosil, P.; Rieseberg, L.H. The speed of ecological speciation. Funct. Ecol. 2007, 21, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunde, J.; Yıldırım, Y.; Tibblin, P.; Bekkevold, D.; Skov, C.; Nordahl, O.; Larsson, P.; Forsman, A. Drivers of neutral and adaptive differentiation in pike (Esox lucius) populations from contrasting environments. Mol. Ecol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.J.; Bradburd, G.S. Isolation by environment. Mol. Ecol. 2014, 23, 5649–5662. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Chen, X.; Wang, J.; Zhou, L.; Zhang, Y.; Zhang, G.; Lu, G.; Wang, C. An integrated and comprehensive transcriptome reveals immune-related genes and signal pathways in topmouth culter (Culter alburnus). Aquac. Res. 2017, 48, 2231–2242. [Google Scholar] [CrossRef]
- Sun, N.; Zhu, D.-M.; Li, Q.; Wang, G.-Y.; Chen, J.; Zheng, F.; Li, P.; Sun, Y.-H. Genetic diversity analysis of Topmouth Culter (Culter alburnus) based on microsatellites and D-loop sequences. Environ. Biol. Fish. 2021, 104, 213–228. [Google Scholar] [CrossRef]
- Li, Y.; Truc, T.; Wang, W. Development of polymorphic microsatellite markers in topmouth culter (Culter alburnus). Conserv. Genet. Resour. 2010, 2, 43–46. [Google Scholar] [CrossRef]
- Qi, P.; Qin, J.; Xie, C. Determination of genetic diversity of wild and cultured topmouth culter (Culter alburnus) inhabiting China using mitochondrial DNA and microsatellites. Biochem. Syst. Ecol. 2015, 61, 232–239. [Google Scholar] [CrossRef]
- Liu, D.; Si, L.; Zhang, X.; Yi, H. Research on embryonic and larvae development of Culter alburnus of Xingkai Lake (In Chinese). J. Northeast. Agric. Univ. 2012, 43, 110–116. [Google Scholar]
- Li, C.; Xu, W.; Wang, J.; Yin, J.; Cao, D.; Shi, L. The artificial propagation and observation of embryonic developemnt in Xingkai lake topmouth culter (Erythroculter ilishaeform) is farmed in a pond. J. Dalian Fish. Univ. 2008, 23, 87–91. [Google Scholar]
- Li, W.; Wang, S.; Hu, J.; Tang, C.; Wu, C.; Liu, J.; Ren, L.; Sun, C.; Dong, J.; Liu, S.; et al. Asymmetric expression of homoeologous genes contributes to dietary adaption of an allodiploid hybrid fish derived from Megalobrama amblycephala (♀) × Culter alburnus (♂). BMC Genomics 2021, 22, 362. [Google Scholar] [CrossRef]
- Salinas, I.; Parra, D. 6-Fish mucosal immunity: Intestine. In Mucosal Health in Aquaculture; Beck, B.H., Peatman, E., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 135–170. [Google Scholar] [CrossRef]
- Le, H.T.M.D.; Shao, X.; Krogdahl, Å.; Kortner, T.M.; Lein, I.; Kousoulaki, K.; Lie, K.K.; Sæle, Ø. Intestinal Function of the Stomachless Fish, Ballan Wrasse (Labrus bergylta). Front. Mar. Sci. 2019, 6, 6. [Google Scholar] [CrossRef]
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. The Gut Microbiota of Marine Fish. Front. Microbiol. 2018, 9, 873. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Chen, W.-D.; Wang, Y.-D. Gut Microbiota: An Integral Moderator in Health and Disease. Front. Microbiol. 2018, 9, 751. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids—At the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Huang, C.; Ge, F.; Yao, X.; Guo, X.; Bao, P.; Ma, X.; Wu, X.; Chu, M.; Yan, P.; Liang, C. Microbiome and Metabolomics Reveal the Effects of Different Feeding Systems on the Growth and Ruminal Development of Yaks. Front. Microbiol. 2021, 12, 1440. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a Prokaryotic Universal Primer for Simultaneous Analysis of Bacteria and Archaea Using Next-Generation Sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Meth. 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, P.; Li, M.; Xie, C.; Xu, L. Chromosome Karyotype analysis of Erythroculter ilishaeformis (In Chinese). J. Hunan Univ. Arts Sci. (Nat. Sci. Ed.) 2009, 21, 73–77. [Google Scholar]
- Lee, J.A.C.; Cossins, A.R. Adaptation of intestinal morphology in the temperature-acclimated carp, Cyprinus carpio L. Cell Tissue Res. 1988, 251, 451–456. [Google Scholar] [CrossRef]
- Chevalier, C.; Stojanović, O.; Colin, D.J.; Suarez-Zamorano, N.; Tarallo, V.; Veyrat-Durebex, C.; Rigo, D.; Fabbiano, S.; Stevanović, A.; Hagemann, S.; et al. Gut Microbiota Orchestrates Energy Homeostasis during Cold. Cell 2015, 163, 1360–1374. [Google Scholar] [CrossRef] [Green Version]
- Alberdi, A.; Aizpurua, O.; Bohmann, K.; Zepeda-Mendoza, M.L.; Gilbert, M.T.P. Do Vertebrate Gut Metagenomes Confer Rapid Ecological Adaptation? Trends Ecol. Evol. 2016, 31, 689–699. [Google Scholar] [CrossRef]
- Smith, C.C.R.; Snowberg, L.K.; Gregory Caporaso, J.; Knight, R.; Bolnick, D.I. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 2015, 9, 2515–2526. [Google Scholar] [CrossRef]
- Naas, A.E.; Mackenzie, A.K.; Mravec, J.; Schückel, J.; Willats, W.G.T.; Eijsink, V.G.H.; Pope, P.B. Do Rumen Bacteroidetes Utilize an Alternative Mechanism for Cellulose Degradation? mBio 2014, 5, e01401–e01414. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Dong, Z.; Cheng, F.; Shi, H.; Zhou, X.; Li, B.; Wang, L.; Wang, W.; Zhang, J. Seasonal diets supersede host species in shaping the distal gut microbiota of Yaks and Tibetan sheep. Sci. Rep. 2021, 11, 22626. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semova, I.; Carten, J.D.; Stombaugh, J.; Mackey, L.C.; Knight, R.; Farber, S.A.; Rawls, J.F. Microbiota Regulate Intestinal Absorption and Metabolism of Fatty Acids in the Zebrafish. Cell Host Microbe 2012, 12, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renouard, S.; Hano, C.; Ouagne, P.; Blondeau, J.-P.; Lainé, E. Protection of flax fiber-based yarns against natural soil degradation by chitosan. Mater. Lett. 2014, 137, 269–273. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiang, M.; Liu, X.; Yu, H.; Otte, M.L.; Ma, C.; Her, Y.G. Environmental variables influencing phytoplankton communities in hydrologically connected aquatic habitats in the Lake Xingkai basin. Ecol. Indic. 2018, 91, 1–12. [Google Scholar] [CrossRef]
- Ju, Y.; Sun, X.; Shabani, E.I.; Zhao, Y.; Li, X.; Yu, T.; Yu, H. Relationships between zooplankton biomass and environmental factors of Xiaoxingkai Lake in northeastern China. Environ. Sci. Pollut. R. 2019, 26, 30279–30285. [Google Scholar] [CrossRef]
- Zhao, R.; Song, N.; Guo, X. Using Phytoplankton Distribution to Assess Eutrophication Condition of XingKai Lake. Heilongjiang Environ. J. 2016, 40, 55–57. [Google Scholar]
- Du, C.; Yang, L.; Zhao, Y.; Wu, X.; Xu, M.; Wang, L.; Zhang, W. Temporal and Spatial Variation of Zooplankton Community Structure and Its Relationship with Environmental Factors in Dianshan Lake, Shanghai. Environ. Sci. 2019, 40, 4513–4521. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Sobs | Chao | Ace | Shannon | Simpson | Pielou |
|---|---|---|---|---|---|---|
| XK | 407 ± 55.77 b | 520.93 ± 81.74 b | 529.16 ± 82.3 b | 5.46 ± 1.05 a | 0.9 ± 0.11 a | 0.63 ± 0.12 a |
| DS | 566 ± 194.14 a | 691.35 ± 174.83 a | 713.43 ± 166.88 a | 5.25 ± 1.21 a | 0.89 ± 0.07 a | 0.58 ± 0.12 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Xu, B.; Zhang, Z.; Zhou, L.; Zhang, G.; Zhang, Y.; Wang, C. Comparison of the Intestinal Structure and Intestinal Microbiome between Two Geographically Isolated Populations of Culter alburnus. Animals 2022, 12, 342. https://doi.org/10.3390/ani12030342
Wang J, Xu B, Zhang Z, Zhou L, Zhang G, Zhang Y, Wang C. Comparison of the Intestinal Structure and Intestinal Microbiome between Two Geographically Isolated Populations of Culter alburnus. Animals. 2022; 12(3):342. https://doi.org/10.3390/ani12030342
Chicago/Turabian StyleWang, Jun, Bowen Xu, Zhiyi Zhang, Lu Zhou, Guoqi Zhang, Youliang Zhang, and Chenghui Wang. 2022. "Comparison of the Intestinal Structure and Intestinal Microbiome between Two Geographically Isolated Populations of Culter alburnus" Animals 12, no. 3: 342. https://doi.org/10.3390/ani12030342