
Captivity Shifts Gut Microbiota Communities in White-Lipped Deer (Cervus albirostris)

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Illumina Sequencing
2.3. Statistical Analysis
3. Results
3.1. Summary of High-Throughput Sequencing
3.2. Composition of the Gut Microbiota between Captive and Wild White-Lipped Deer
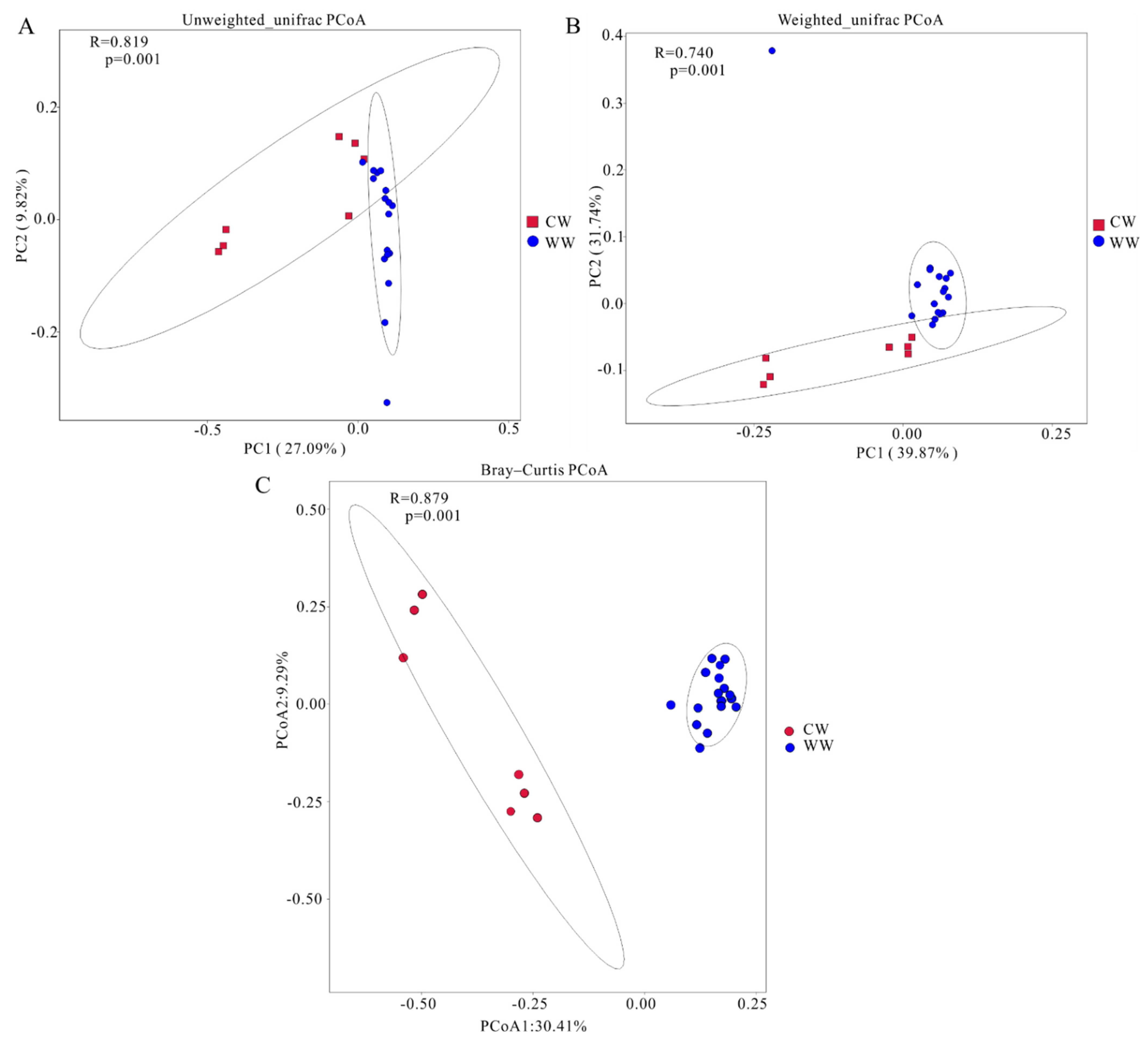
3.3. Variation of Gut Microbiota Diversity across Different Living Environments
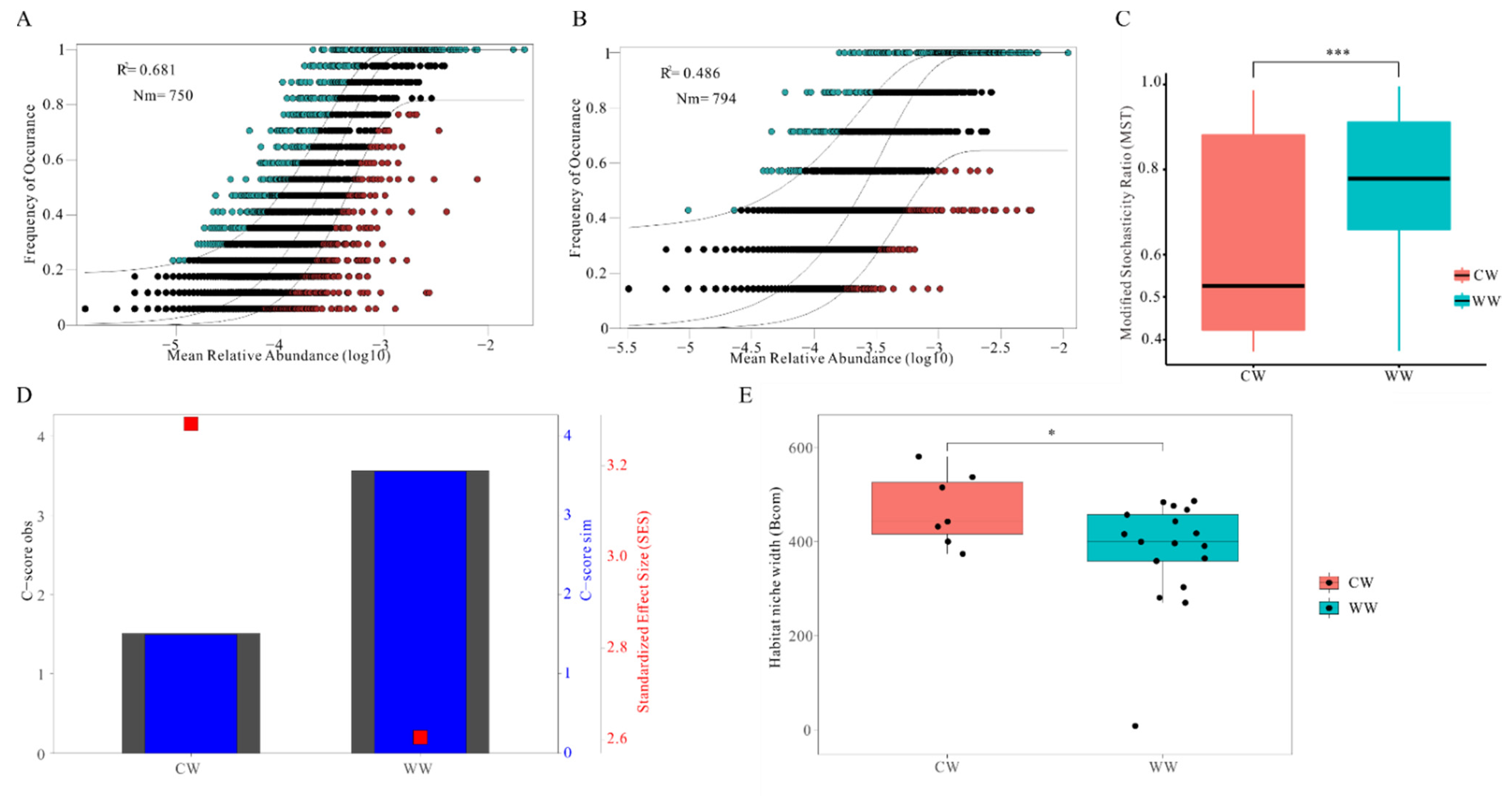
3.4. Relative Importance of Deterministic and Stochastic Processesin CW and WW
4. Discussion
4.1. Captivity Changes the Composition of Gut Microbiota
4.2. Alpha and Beta Diversity in Gut Microbiota
4.3. Captivity Mediates the Ecological Assembly Process of Gut Microbiota Communities
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- You, Z.; Tang, Z.; Yang, Y.; Yang, L.; Shi, H.; Liu, H.; Gan, X.; Zheng, T.; Jiang, Z. Summer habitat selection by white-lipped deer (Cervus albirostris) in Chaqingsongduo White-lipped Deer National Nature Reserve. Acta Theriol. Sin. 2014, 34, 46–53. [Google Scholar] [CrossRef]
- Wei, F.; Yang, Q.; Wu, Y.; Jiang, X.; Liu, S.; Li, B.; Yang, G.; Li, M.; Zhou, J.; Li, S.; et al. Catalogue of mammals in China (2021). Acta Theriol. Sin. 2021, 41, 487–501. [Google Scholar] [CrossRef]
- Cui, S.; Luo, X.; Li, C.; Hu, H.; Jiang, Z. Predicting the potential distribution of white-lipped deer using the MaxEnt model. Biodivers. Sci. 2018, 26, 171–176. [Google Scholar] [CrossRef]
- Kaji, K.; Ohtaishi, N.; Miura, S.; Wu, J. Distribution and status of White-lipped Deer (Cervus albirostris) in the Qinghai-Xizang (Tibet) Plateau, China. Mammal Rev. 1989, 19, 35–44. [Google Scholar] [CrossRef]
- Jiayan, W.; Junfeng, F. Present Status of Research of White-lipped Deer and its Conservation Strategy. Chin. J. Wildl. 2007, 5, 36–39. [Google Scholar] [CrossRef]
- Hughes, J.; Alexander, J.; Shi, K.; Riordan, P. Confirmation of threatened white-lipped deer (Przewalskium albirostris) in Gansu and Sichuan, China, and their overlap with livestock. Mammalia 2015, 79, 241–244. [Google Scholar] [CrossRef]
- Harris, R.; Brook, S.M.; McShea, W. Cervus Albirostris: The IUCN Red List of Threatened Species 2015: E. T4256A61976756. IUCN Red List Threat Species. 2015. Available online: https://doi.org/10.2305/IUCN.UK.2015-2.RLTS.T4256A61976756.en (accessed on 2 November 2021).
- Jiang, Z.; Jiang, J.; Wang, Y.; Zhang, E.; Zhang, Y.; Li, L.; Xie, F.; Cai, B.; Cao, L.; Zheng, G. Red list of China’s vertebrates. Biodivers. Sci. 2016, 24, 500–551. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [Green Version]
- Rosa, E.; Minard, G.; Lindholm, J.; Saastamoinen, M. Moderate plant water stress improves larval development and impacts immunity and gut microbiota of a specialist herbivore. PLoS ONE 2019, 14, e0204292. [Google Scholar] [CrossRef]
- Brune, A.; Ohkuma, M. Role of the termite gut microbiota in symbiotic digestion. In Biology of Termites: A Modern Synthesis; Bignell, D.E., Roisin, Y., Lo, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 439–475. [Google Scholar]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Everard, A.; Duparc, T. Gut microbiota, enteroendocrine functions and metabolism. Curr. Opin. Pharmacol. 2013, 13, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Dearing, M.D.; Kohl, K.D. Beyond fermentation: Other important services provided to endothermic herbivores by their gut microbiota. Integr. Comp. Biol. 2017, 57, 723–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, B.; Wright, A.-D. Diversity of gut methanogens in herbivorous animals. Animal 2013, 7, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhang, M.; Wang, S.; Han, R.; Cao, Y.; Hua, W.; Mao, Y.; Zhang, X.; Pang, X.; Wei, C.; et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 2010, 4, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Liu, W.; Tao, H.; Zhang, Y.; Liu, L.; Liu, Z.; Qiu, B.; Xu, T. Effect of industrial trans-fatty acids-enriched diet on gut microbiota of C57BL/6 mice. Eur. J. Nutr. 2019, 58, 2625–2638. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Zhang, T.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; et al. Comparative analysis of the gut microbiota composition between captive and wild forest musk deer. Front. Microbiol. 2017, 8, 1705. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Mishra, S.; Wang, C.; Zhang, H.; Ning, R.; Kong, F.; Zeng, B.; Zhao, J.; Li, Y. Comparative study of gut microbiota in wild and captive giant pandas (Ailuropoda melanoleuca). Genes 2019, 10, 827. [Google Scholar] [CrossRef] [Green Version]
- Menke, S.; Melzheimer, J.; Thalwitzer, S.; Heinrich, S.; Wachter, B.; Sommer, S. Gut microbiomes of free-ranging and captive Namibian cheetahs: Diversity, putative functions and occurrence of potential pathogens. Mol. Ecol. 2017, 26, 5515–5527. [Google Scholar] [CrossRef]
- Liu, C.; Hu, J.; Wu, Y.; Irwin, D.M.; Chen, W.; Zhang, Z.; Yu, L. Comparative study of gut microbiota from captive and confiscated-rescued wild pangolins. J. Genet. Genom. 2021, 48, 825–835. [Google Scholar] [CrossRef]
- Li, J.; Wang, C.; Tang, Z.; Guo, Y.; Zheng, T.; Li, Y.; You, Z. The gut bacterial community composition of wild Cervus albirostris (white-lipped deer) detected by the 16S ribosomal RNA gene sequencing. Curr. Microbiol. 2017, 74, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ning, D. Stochastic community assembly: Does it matter in microbial ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002–e00017. [Google Scholar] [CrossRef] [Green Version]
- Hubbell, S. Neutral theory in community ecology and the hypothesis of functional equivalence. Funct. Ecol. 2005, 19, 166–172. [Google Scholar] [CrossRef]
- Stegen, J.C.; Fredrickson, J.K.; Wilkins, M.J.; Konopka, A.E.; Nelson, W.C.; Arntzen, E.V.; Chrisler, W.B.; Chu, R.K.; Danczak, R.E.; Fansler, S.J.; et al. Groundwater–surface water mixing shifts ecological assembly processes and stimulates organic carbon turnover. Nat. Commun. 2016, 7, 11237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, E.B.; Crump, A.R.; Resch, C.T.; Fansler, S.; Arntzen, E.; Kennedy, D.W.; Fredrickson, J.K.; Stegen, J.C. Deterministic influences exceed dispersal effects on hydrologically-connected microbiomes. Environ. Microbiol. 2017, 19, 1552–1567. [Google Scholar] [CrossRef] [PubMed]
- Graham, E.B.; Crump, A.R.; Resch, C.T.; Fansler, S.; Arntzen, E.; Kennedy, D.W.; Fredrickson, J.K.; Stegen, J.C. Coupling spatiotemporal community assembly processes to changes in microbial metabolism. Front. Microbiol. 2016, 7, 1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, I.; Maldonado-Gomez, M.X.; Gomes-Neto, J.C.; Kittana, H.; Ding, H.; Schmaltz, R.; Joglekar, P.; Cardona, R.J.; Marsteller, N.L.; Kembel, S.W.; et al. Experimental evaluation of the importance of colonization history in early-life gut microbiota assembly. Elife 2018, 7, e36521. [Google Scholar] [CrossRef]
- Li, H.; Zhou, R.; Zhu, J.; Huang, X.; Qu, J. Environmental filtering increases with elevation for the assembly of gut microbiota in wild pikas. Microb. Biotechnol. 2019, 12, 976–992. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. bioRxiv 2020, 5, 326–504. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Oksanen, J. Multivariate analysis of ecological communities in R: Vegan tutorial. R Package Version 2011, 1, 1–43. [Google Scholar]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, D.; Deng, Y.; Tiedje, J.M.; Zhou, J. A general framework for quantitatively assessing ecological stochasticity. Proc. Natl. Acad. Sci. USA 2019, 116, 16892–16898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, L.; Roberts, A. The checkerboard score and species distributions. Oecologia 1990, 85, 74–79. [Google Scholar] [CrossRef]
- Mo, Y.; Peng, F.; Gao, X.; Xiao, P.; Logares, R.; Jeppesen, E.; Ren, K.; Xue, Y.; Yang, J. Low shifts in salinity determined assembly processes and network stability of microeukaryotic plankton communities in a subtropical urban reservoir. Microbiome 2021, 9, 128. [Google Scholar] [CrossRef]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Pandit, S.N.; Kolasa, J.; Cottenie, K. Contrasts between habitat generalists and specialists: An empirical extension to the basic metacommunity framework. Ecology 2009, 90, 2253–2262. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Yang, H.; Han, S.; Feng, L.; Wang, T.; Ge, J. Comparison of the gut microbiota composition between wild and captive sika deer (Cervus nippon hortulorum) from feces by high-throughput sequencing. AMB Express 2017, 7, 212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, X.; Gao, H.; Wu, G.; Qin, W.; Song, P.; Wang, L.; Chen, J.; Cai, Z.; Zhang, T. Comparison of gut microbiota diversity between wild and captive bharals (Pseudois nayaur). BMC Vet. Res. 2019, 15, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Chi, X.; Qin, W.; Wang, L.; Song, P.; Cai, Z.; Zhang, J.; Zhang, T. Comparison of the gut microbiota composition between the wild and captive Tibetan wild ass (Equus kiang). J. Appl. Microbiol. 2019, 126, 1869–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D. The emerging view of Firmicutes as key fibre degraders in the human gut. Environ. Microbiol. 2016, 18, 2081–2083. [Google Scholar] [CrossRef]
- Bernini, L.J.; Simão, A.N.C.; Alfieri, D.F.; Lozovoy, M.A.B.; Mari, N.L.; de Souza, C.H.B.; Dichi, I.; Costa, G.N. Beneficial effects of Bifidobacterium lactis on lipid profile and cytokines in patients with metabolic syndrome: A randomized trial. Effects of probiotics on metabolic syndrome. Nutrition 2016, 32, 716–719. [Google Scholar] [CrossRef]
- Martínez, I.; Lattimer, J.M.; Hubach, K.L.; Case, J.A.; Yang, J.; Weber, C.G.; Louk, J.A.; Rose, D.J.; Kyureghian, G.; Peterson, D.A.; et al. Gut microbiome composition is linked to whole grain-induced immunological improvements. ISME J. 2013, 7, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Dolfing, J.; Wang, B.; Chen, R.; Huang, M.; Li, Z.; Lin, X.; Feng, Y. Bacterial communities involved directly or indirectly in the anaerobic degradation of cellulose. Biol. Fertil. Soils 2019, 55, 201–211. [Google Scholar] [CrossRef]
- Gavande, P.V.; Basak, A.; Sen, S.; Lepcha, K.; Murmu, N.; Rai, V.; Mazumdar, D.; Saha, S.P.; Das, V.; Ghosh, S. Functional characterization of thermotolerant microbial consortium for lignocellulolytic enzymes with central role of Firmicutes in rice straw depolymerization. Sci. Rep. 2021, 11, 3032. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.-R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Bay, S.K.; Dong, X.; Bradley, J.A.; Leung, P.M.; Grinter, R.; Jirapanjawat, T.; Arndt, S.K.; Cook, P.L.; LaRowe, D.E.; Nauer, P.; et al. Trace gas oxidizers are widespread and active members of soil microbial communities. Nat. Microbiol. 2021, 6, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.C.; Healey, G.R.; Kelly, W.J.; Patchett, M.L.; Jordens, Z.; Tannock, G.W.; Sims, I.M.; Bell, T.J.; Hedderley, D.; Henrissat, B.; et al. Genomic insights from Monoglobus pectinilyticus: A pectin-degrading specialist bacterium in the human colon. ISME J. 2019, 13, 1437–1456. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.; Rodriguez-Palacios, A. The genus Alistipes: Gut bacteria with emerging implications to inflammation, cancer, and mental health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; Angelis, M.D. The controversial role of human gut. Lachnospiraceae. Microorg. 2020, 8, 573. [Google Scholar] [CrossRef]
- Tavella, T.; Rampelli, S.; Guidarelli, G.; Bazzocchi, A.; Gasperini, C.; Pujos-Guillot, E.; Comte, B.; Barone, M.; Biagi, E.; Candela, M.; et al. Elevated gut microbiome abundance of Christensenellaceae, Porphyromonadaceae and Rikenellaceae is associated with reduced visceral adipose tissue and healthier metabolic profile in Italian elderly. Gut Microbes 2021, 13, 1–19. [Google Scholar] [CrossRef]
- Gao, H.; Chi, X.; Li, G.; Qin, W.; Song, P.; Jiang, F.; Liu, D.; Zhang, J.; Zhou, X.; Li, S.; et al. Gut microbial diversity and stabilizing functions enhance the plateau adaptability of Tibetan wild ass (Equus kiang). MicrobiologyOpen 2020, 9, 1150–1161. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Xia, T.; Wei, Q.; Dong, Y.; Zhao, C.; Yang, X.; Zhang, L.; Wang, X.; Sha, W.; Zhang, H. Analysis of gut microbiota in three species belonging to different genera (Hemitragus, Pseudois, and Ovis) from the subfamily Caprinae in the absence of environmental variance. Ecol. Evol. 2021, 11, 12129–12140. [Google Scholar] [CrossRef]
- Shang, Z.; Kong, Q.; Li, J.; Liu, S.; Tan, Z.; Shang, P.; Wang, H. Characterization of Bacterial Microbial Diversity in Wild Yak and Domestic Yak in Qiangtang Region of Tibet. Pak. J. Zool. 2021, 54, 1001–1009. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, M.; Fan, M.; Xu, S.; Li, Y.; Zhang, T.; Cha, M.; Liu, Y.; Guo, X.; Chen, Q.; et al. Comparative analysis of gut microbiota changes in Père David’s deer populations in Beijing Milu Park and Shishou, Hubei Province in China. Front. Microbiol. 2018, 9, 1258. [Google Scholar] [CrossRef]
- Menke, S.; Heurich, M.; Henrich, M.; Wilhelm, K.; Sommer, S. Impact of winter enclosures on the gut bacterial microbiota of red deer in the Bavarian Forest National Park. Wildl. Biol. 2019, 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Minich, D.; Madden, C.; Evans, M.V.; Ballash, G.A.; Barr, D.J.; Poulsen, K.P.; Dennis, P.M.; Hale, V.L. Alterations in gut microbiota linked to provenance, sex, and chronic wasting disease in white-tailed deer (Odocoileus virginianus). Sci. Rep. 2021, 11, 13218. [Google Scholar] [CrossRef]
- Liu, L.; Hou, J.; Yu, W.; Cao, F.; Li, K.; Wu, Q.; Zhang, J. Study on the Histology of Gastrointestine of White-Lipped Deer (Cervus albirostrine). J. Yangzhou Univ. 1987, 8, 37–40. [Google Scholar] [CrossRef]
- Dini-Andreote, F.; Stegen, J.C.; Van Elsas, J.D.; Salles, J.F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Shi, Y.; Cui, X.; Yue, P.; Li, K.; Liu, X.; Tripathi, B.M.; Chu, H.Y. Salinity is a key determinant for soil microbial communities in a desert ecosystem. Msystems 2019, 4, e00218–e00225. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, B.; Liu, Y.; Guo, Y.; Shi, P.; Wei, G.H. Distinct large-scale biogeographic patterns of fungal communities in bulk soil and soybean rhizosphere in China. Sci. Total Environ. 2018, 644, 791–800. [Google Scholar] [CrossRef]
- Chen, W.; Ren, K.; Isabwe, A.; Chen, H.; Liu, M.; Yang, J. Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 2019, 7, 138. [Google Scholar] [CrossRef] [Green Version]
- Carmody, R.N.; Gerber, G.K.; Luevano Jr, J.M.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Nelson, T.M.; Rogers, T.L.; Carlini, A.R.; Brown, M.V. Diet and phylogeny shape the gut microbiota of a ntarctic seals: A comparison of wild and captive animals. Environ. Microbiol. 2013, 15, 1132–1145. [Google Scholar] [CrossRef]
- Doré, J.; Blottière, H. The influence of diet on the gut microbiota and its consequences for health. Curr. Opin. Biotechnol. 2015, 32, 195–199. [Google Scholar] [CrossRef]
- Martínez-Mota, R.; Kohl, K.D.; Orr, T.J.; Dearing, M.D. Natural diets promote retention of the native gut microbiota in captive rodents. ISME J. 2020, 14, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Dearing, M.D. Wild-caught rodents retain a majority of their natural gut microbiota upon entrance into captivity. Environ. Microbiol. Rep. 2014, 6, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.K.; McKenney, E.A.; O’Connell, T.M.; Drea, C.M. The critical role of dietary foliage in maintaining the gut microbiome and metabolome of folivorous sifakas. Sci. Rep. 2018, 8, 14482. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.L.; Wiley, N.; Cooke, A.C.; Johnson, C.N.; Fouhy, F.; Reichert, M.S.; de la Hera, I.; Crane, J.M.; Kulahci, I.G.; Ross, R.P.; et al. Diet induces parallel changes to the gut microbiota and problem solving performance in a wild bird. Sci. Rep. 2020, 10, 20783. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Gao, H.; Song, P.; Liang, C.; Jiang, F.; Xu, B.; Liu, D.; Zhang, T. Captivity Shifts Gut Microbiota Communities in White-Lipped Deer (Cervus albirostris). Animals 2022, 12, 431. https://doi.org/10.3390/ani12040431
Li B, Gao H, Song P, Liang C, Jiang F, Xu B, Liu D, Zhang T. Captivity Shifts Gut Microbiota Communities in White-Lipped Deer (Cervus albirostris). Animals. 2022; 12(4):431. https://doi.org/10.3390/ani12040431
Chicago/Turabian StyleLi, Bin, Hongmei Gao, Pengfei Song, Chenbo Liang, Feng Jiang, Bo Xu, Daoxin Liu, and Tongzuo Zhang. 2022. "Captivity Shifts Gut Microbiota Communities in White-Lipped Deer (Cervus albirostris)" Animals 12, no. 4: 431. https://doi.org/10.3390/ani12040431
APA StyleLi, B., Gao, H., Song, P., Liang, C., Jiang, F., Xu, B., Liu, D., & Zhang, T. (2022). Captivity Shifts Gut Microbiota Communities in White-Lipped Deer (Cervus albirostris). Animals, 12(4), 431. https://doi.org/10.3390/ani12040431