
Ocular Microbiome in a Group of Clinically Healthy Horses

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Subjects and Inclusion Criteria
2.3. DNA Extraction, Targeted Sequencing and Bioinformatic Analysis
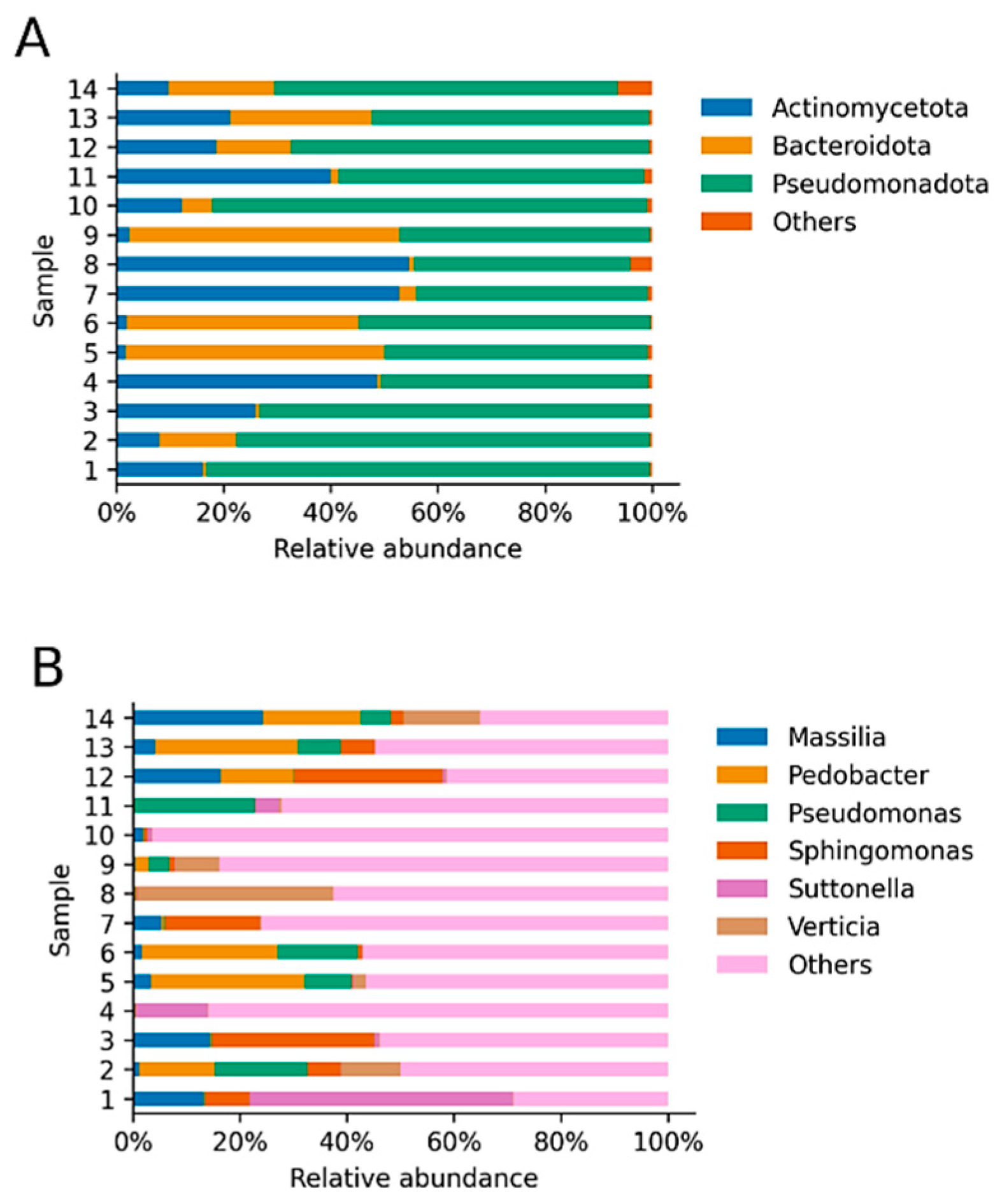
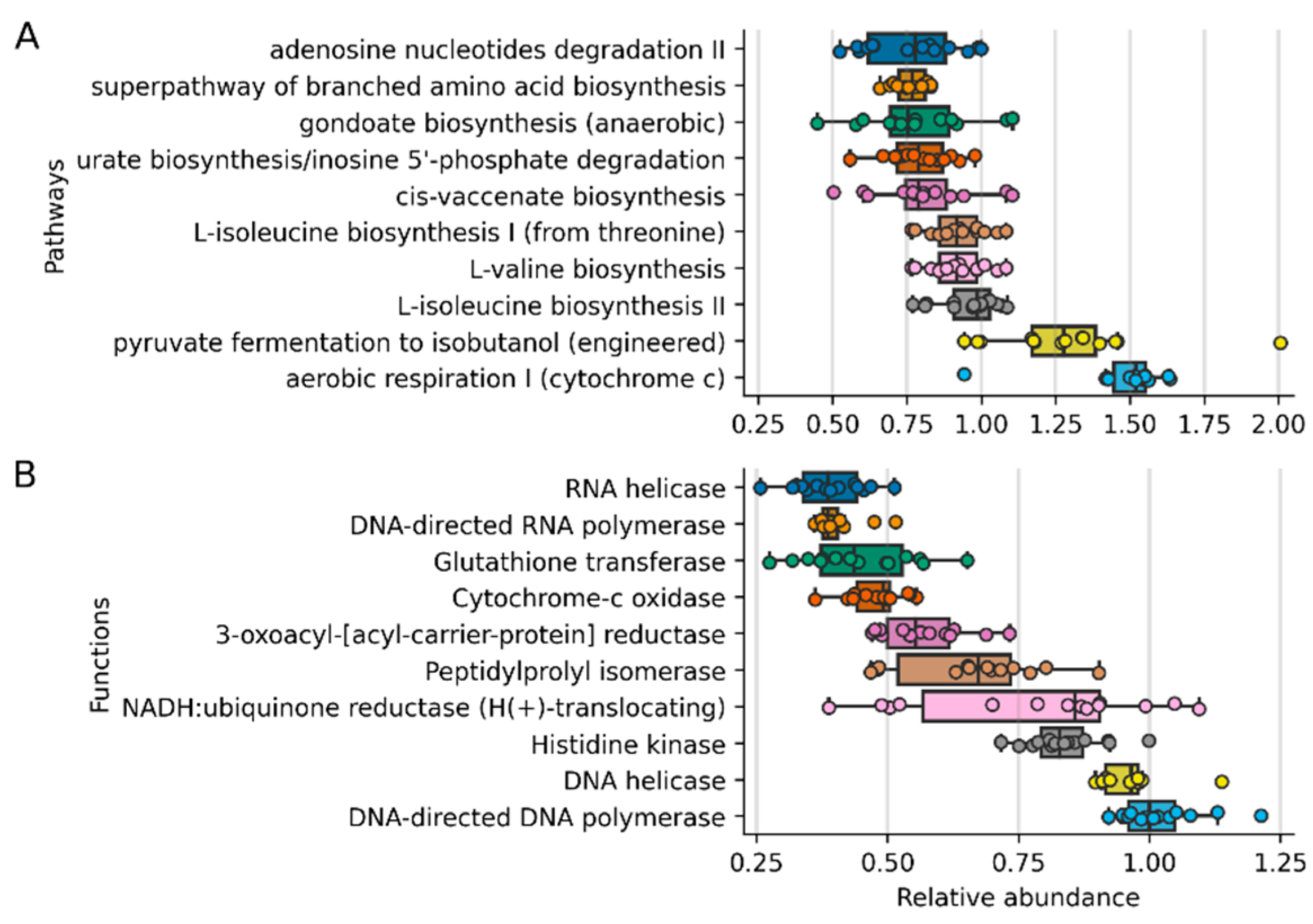
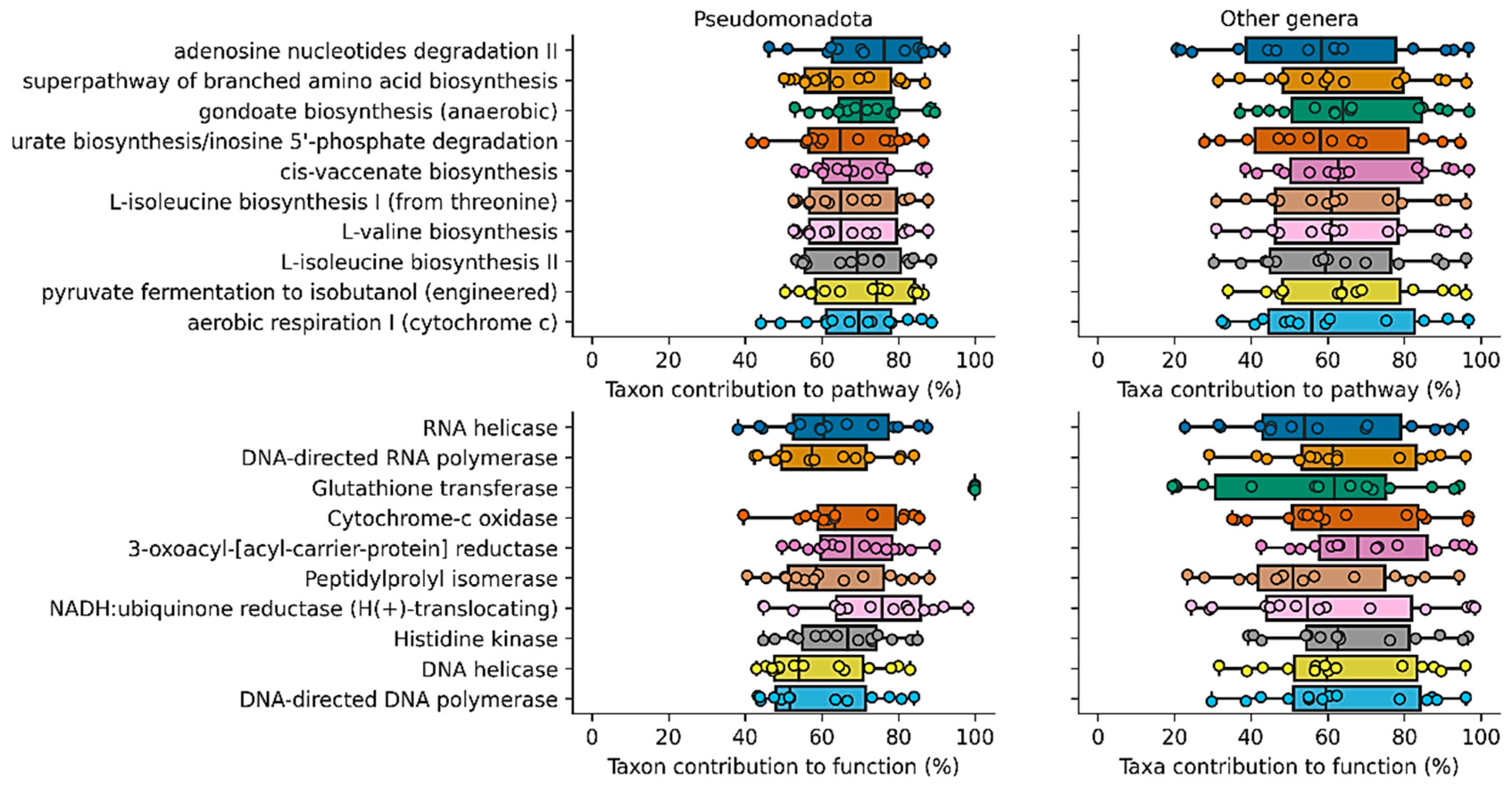
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kugadas, A.; Gadjeva, M. Impact of Microbiome on Ocular Health. Ocul. Surf. 2016, 14, 342–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozkan, J.; Willcox, M.D. The Ocular Microbiome: Molecular Characterization of a Unique and Low Microbial Environment. Curr. Eye Res. 2019, 44, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Diez-Vives, C.; Coroneo, M.; Thomas, T.; Willcox, M. Temporal Stability and Composition of the Ocular Surface Microbiome. Sci. Rep. 2017, 7, 9880. [Google Scholar] [CrossRef] [Green Version]
- Aragona, P.; Baudouin, C.; Del Castillo, J.M.; Messmer, E.; Barabino, S.; Merayo-Lloves, J.; Brignole-Baudouin, F.; Inferrera, L.; Rolando, M.; Mencucci, R.; et al. The ocular microbiome and microbiota and their effects on ocular surface pathophysiology and disorders. Surv. Ophthalmol. 2021, 66, 907–925. [Google Scholar] [CrossRef]
- Willcox, M.D.; Argüeso, P.; Georgiev, G.; Holopainen, J.M.; Laurie, G.; Millar, T.J.; Papas, E.B.; Rolland, J.P.; Schmidt, T.A.; Stahl, U.; et al. TFOS DEWS II Tear Film Report. Ocul. Surf. 2017, 15, 366–403. [Google Scholar] [CrossRef] [Green Version]
- Leger, A.J.S.; Desai, J.; Drummond, R.; Kugadas, A.; Almaghrabi, F.; Silver, P.; Raychaudhuri, K.; Gadjeva, M.; Iwakura, Y.; Lionakis, M.S.; et al. An Ocular Commensal Protects against Corneal Infection by Driving an Interleukin-17 Response from Mucosal γδ T Cells. Immunity 2017, 47, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Borroni, D.; Romano, V.; Kaye, S.B.; Somerville, T.; Napoli, L.; Fasolo, A.; Gallon, P.; Ponzin, D.; Esposito, A.; Ferrari, S. Metagenomics in ophthalmology: Current findings and future prospectives. BMJ Open Ophthalmol. 2019, 4, e000248. [Google Scholar] [CrossRef]
- Gomes, J.Á.P.; Frizon, L.; Demeda, V.F. Ocular surface microbiome in health and disease. Asia Pac. J. Ophthalmol. 2020, 9, 505–511. [Google Scholar] [CrossRef]
- Scott, E.M.; Arnold, C.; Dowell, S.; Suchodolski, J.S. Evaluation of the bacterial ocular surface microbiome in clinically normal horses before and after treatment with topical neomycin-polymyxin-bacitracin. PLoS ONE 2019, 14, e0214877. [Google Scholar] [CrossRef] [Green Version]
- LaFrentz, B.R.; LaFrentz, S.A.; Beck, B.H.; Arias, C.R. Draft genome sequences of Cetobacterium somerae 2G large and two novel Cetobacterium isolates from intestines of channel catfish (Ictalurus punctatus). Microbiol. Resour. Announc. 2020, 9, e1006–e1020. [Google Scholar] [CrossRef]
- Rogers, C.M.; Scott, E.M.; Sarawichitr, B.; Arnold, C.; Suchodolski, J.S. Evaluation of the bacterial ocular surface microbiome in ophthalmologically normal dogs prior to and following treatment with topical neomycin-polymyxin-bacitracin. PLoS ONE 2020, 15, e0234313. [Google Scholar] [CrossRef]
- Walsh, M.L.; Meason-Smith, C.; Arnold, C.; Suchodolski, J.S.; Scott, E.M. Evaluation of the ocular surface mycobiota in clinically normal horses. PLoS ONE 2021, 16, e0246537. [Google Scholar] [CrossRef]
- Dicks, L.M.T.; Mikkelsen, L.S.; Brandsborg, E.; Marcotte, H. Clostridium difficile, the Difficult ‘Kloster’ Fuelled by Antibiotics. Curr. Microbiol. 2018, 76, 774–782. [Google Scholar] [CrossRef]
- Cavuoto, K.M.; Banerjee, S.; Galor, A. Relationship between the microbiome and ocular health. Ocul. Surf. 2019, 17, 384–392. [Google Scholar] [CrossRef]
- Okonkwo, A.; Rimmer, V.; Walkden, A.; Brahma, A.; Carley, F.; McBain, A.J.; Radhakrishnan, H. Next-Generation Sequencing of the Ocular Surface Microbiome: In Health, Contact Lens Wear, Diabetes, Trachoma, and Dry Eye. Eye Contact Lens Sci. Clin. Pract. 2020, 46, 254–261. [Google Scholar] [CrossRef]
- Zilliox, M.J.; Gange, W.; Kuffel, G.; Mores, C.R.; Joyce, C.; de Bustros, P.; Bouchard, C.S. Assessing the ocular surface microbiome in severe ocular surface diseases. Ocul. Surf. 2020, 18, 706–712. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Beli, E.; Yan, Y.; Moldovan, L.; Vieira, C.P.; Gao, R.; Duan, Y.; Prasad, R.; Bhatwadekar, A.; White, F.A.; Townsend, S.D.; et al. Restructuring of the gut microbiome by intermittent fasting prevents retinopathy and prolongs survival in db/db mice. J. Diabetes 2018, 67, 1867–1879. [Google Scholar] [CrossRef] [Green Version]
- Vicencio, J. Boletín S2S—Pronóstico Subestacional y Estacional. Oficina Servicios Climáticos Sección Climatología Dirección Meteorológica de Chile No 164. Available online: http://www.meteochile.gob.cl (accessed on 1 December 2020).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Thomson, P.; Santibañez, R.; Aguirre, C.; Galgani, J.E.; Garrido, D. Short-term impact of sucralose consumption on the metabolic response and gut microbiome of healthy adults. Br. J. Nutr. 2019, 122, 856–862. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “all-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2013, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Oren, A.; Arahal, D.R.; Rosselló-Móra, R.; Sutcliffe, I.C.; Moore, E.R.B. Emendation of Rules 5b, 8, 15 and 22 of the International Code of Nomenclature of Prokaryotes to include the rank of phylum. Int. J. Syst. Evol. Microbiol. 2021, 71, 004851. [Google Scholar] [CrossRef]
- Lloyd, K.G.; Tahon, G. Science depends on nomenclature, but nomenclature is not science. Nat. Rev. Genet. 2022, 20, 123–124. [Google Scholar] [CrossRef]
- LaFrentz, S.; Abarca, E.; Mohammed, H.H.; Cuming, R.; Arias, C.R. Characterization of the normal equine conjunctival bacterial community using culture-independent methods. Vet. Ophthalmol. 2020, 23, 480–488. [Google Scholar] [CrossRef]
- Lu, L.J.; Liu, J. Human Microbiota and Ophthalmic Disease. Yale J. Biol. Med. 2016, 89, 325–330. [Google Scholar]
- Leis, M.L.; Costa, M.O. Initial description of the core ocular surface microbiome in dogs: Bacterial community diversity and composition in a defined canine population. Vet. Ophthalmol. 2019, 22, 337–344. [Google Scholar] [CrossRef]
- Weese, S.J.; Nichols, J.; Jalali, M.; Litster, A. The oral and conjunctival microbiotas in cats with and without feline immunodeficiency virus infection. Vet. Res. 2015, 46, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Płoneczka-Janeczko, K.; Bania, J.; Bierowiec, K.; Kiełbowicz, M.; Kiełbowicz, Z. Bacterial Diversity in Feline Conjunctiva Based on 16S rRNA Gene Sequence Analysis: A Pilot Study. Biomed Res. Int. 2017, 2017, 3710404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.; Apel, A.; Stapleton, F. Risk Factors and Causative Organisms in Microbial Keratitis at the Princess Alexandra. Cornea 2008, 27, 22–27. [Google Scholar] [CrossRef]
- Sauer, P.; Andrew, S.E.; Lassaline, M.; Gelatt, K.N.; Denis, H.M. Changes in Antibiotic Resistance in Equine Bacterial Ulcerative Keratitis (1991–2000): 65 Horses. Vet. Ophthalmol. 2003, 6, 309–313. [Google Scholar] [CrossRef]
- Wada, S.; Hobo, S.; Niwa, H.; Wada, S. Ulcerative Keratitis in Thoroughbred Racehorses in Japan from 1997 to 2008. Vet. Ophthalmol. 2010, 13, 99–105. [Google Scholar] [CrossRef]
- Kämpfer, P.; Lodders, N.; Martin, K.; Falsen, E. Massilia oculi sp. nov., isolated from a human clinical specimen. Int. J. Syst. Evol. Microbiol. 2012, 62, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Wang, S.; Shen, J.; Zhu, B. Complete Genome Sequence of Massilia oculi sp. nov. CCUG 43427T (=DSM 26321T), the Type Strain of M. oculi, and Comparison with Genome Sequences of Other Massilia Strains. Curr. Microbiol. 2018, 76, 1082–1086. [Google Scholar] [CrossRef]
- Johns, I.C.; Baxter, K.; Booler, H.; Hicks, C.; Menzies-Gow, N. Conjunctival Bacterial and Fungal Flora in Healthy Horses in the UK. Vet. Ophthalmol. 2011, 14, 195–199. [Google Scholar] [CrossRef]
- Hampson, E.C.G.M.; Gibson, J.S.; Barot, M.; Shapter, F.; Greer, R.M. Identification of bacteria and fungi sampled from the conjunctival surface of normal horses in South-East Queensland, Australia. Vet. Ophthalmol. 2018, 22, 265–275. [Google Scholar] [CrossRef]
- Andrew, S.E.; Nguyen, A.; Jones, G.L.; Brooks, D.E. Seasonal Effects on the Aerobic Bacterial and Fungal Conjunctival Flora of Normal Thoroughbred Brood Mares in Florida. Vet. Ophthalmol. 2003, 6, 45–50. [Google Scholar] [CrossRef]
- Zak, A.; Siwinska, N.; Slowikowska, M.; Borowicz, H.; Płoneczka-Janeczko, K.; Chorbiński, P.; Niedzwiedz, A. Conjunctival aerobic bacterial flora in healthy Silesian foals and adult horses in Poland. BMC Vet. Res. 2018, 14, 261. [Google Scholar] [CrossRef] [Green Version]
- Jinks, M.R.; Miller, E.J.; Diaz-Campos, D.; Mollenkopf, D.F.; Newbold, G.; Gemensky-Metzler, A.; Chandler, H.L. Using minimum inhibitory concentration values of common topical antibiotics to investigate emerging antibiotic resistance: A retrospective study of 134 dogs and 20 horses with ulcerative keratitis. Vet. Ophthalmol. 2020, 23, 806–813. [Google Scholar] [CrossRef]
- Bourély, C.; Cazeau, G.; Jarrige, N.; Haenni, M.; Gay, E.; Leblond, A. Antimicrobial resistance in bacteria isolated from diseased horses in France. Equine Vet. J. 2019, 52, 112–119. [Google Scholar] [CrossRef]
- Papalia, M.; Steffanowski, C.; Traglia, G.M.; Almuzara, M.; Martina, P.; Galanternik, L.; Vay, C.; Gutkind, G.; Ramírez, M.S.; Radice, M. Diversity of Achromobacter species recovered from patients with cystic fibrosis, in Argentina. Rev. Argent. Microbiol. 2020, 52, 13–18. [Google Scholar] [CrossRef]
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef]
- Yamamoto, K.; Tsuchisaka, A.; Yukawa, H. Branched-Chain amino acids. Adv. Biochem. Eng. Biotechnol. 2017, 159, 103–128. [Google Scholar] [CrossRef]
- Zheng, X.; Cui, Y.; Li, T.; Li, R.; Guo, L.; Li, D.; Wu, B. Biochemical and structural characterization of a highly active branched-chain amino acid aminotransferase from Pseudomonas sp. for efficient biosynthesis of chiral amino acids. Appl. Microbiol. Biotechnol. 2019, 103, 8051–8062. [Google Scholar] [CrossRef]
- Patakova, P.; Kolek, J.; Sedlar, K.; Koscova, P.; Branska, B.; Kupkova, K.; Paulova, L.; Provaznik, I. Comparative analysis of high butanol tolerance and production in clostridia. Biotechnol. Adv. 2018, 36, 721–738. [Google Scholar] [CrossRef]
- Liao, C.; Wang, T.; Maslov, S.; Xavier, J.B. Modeling microbial cross-feeding at intermediate scale portrays community dynamics and species coexistence. PLoS Comput. Biol. 2020, 16, e1008135. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santibáñez, R.; Lara, F.; Barros, T.M.; Mardones, E.; Cuadra, F.; Thomson, P. Ocular Microbiome in a Group of Clinically Healthy Horses. Animals 2022, 12, 943. https://doi.org/10.3390/ani12080943
Santibáñez R, Lara F, Barros TM, Mardones E, Cuadra F, Thomson P. Ocular Microbiome in a Group of Clinically Healthy Horses. Animals. 2022; 12(8):943. https://doi.org/10.3390/ani12080943
Chicago/Turabian StyleSantibáñez, Rodrigo, Felipe Lara, Teresa M. Barros, Elizabeth Mardones, Françoise Cuadra, and Pamela Thomson. 2022. "Ocular Microbiome in a Group of Clinically Healthy Horses" Animals 12, no. 8: 943. https://doi.org/10.3390/ani12080943
APA StyleSantibáñez, R., Lara, F., Barros, T. M., Mardones, E., Cuadra, F., & Thomson, P. (2022). Ocular Microbiome in a Group of Clinically Healthy Horses. Animals, 12(8), 943. https://doi.org/10.3390/ani12080943