
The Multi-Omics Analysis Revealed a Metabolic Regulatory System of Cecum in Rabbit with Diarrhea

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Feeding Conditions
2.3. Sample Collection
2.4. Pathological Analysis
2.5. DNA Extraction, 16S rRNA Sequencing, and Sequence Analysis
2.6. Transcriptome Sequencing and Differentially Expressed Genes Analysis
2.7. UHPLC-MS/MS-Based Non-Targeted Metabolomics Analysis
2.8. Association Analysis
2.9. Statistical Analysis
3. Results
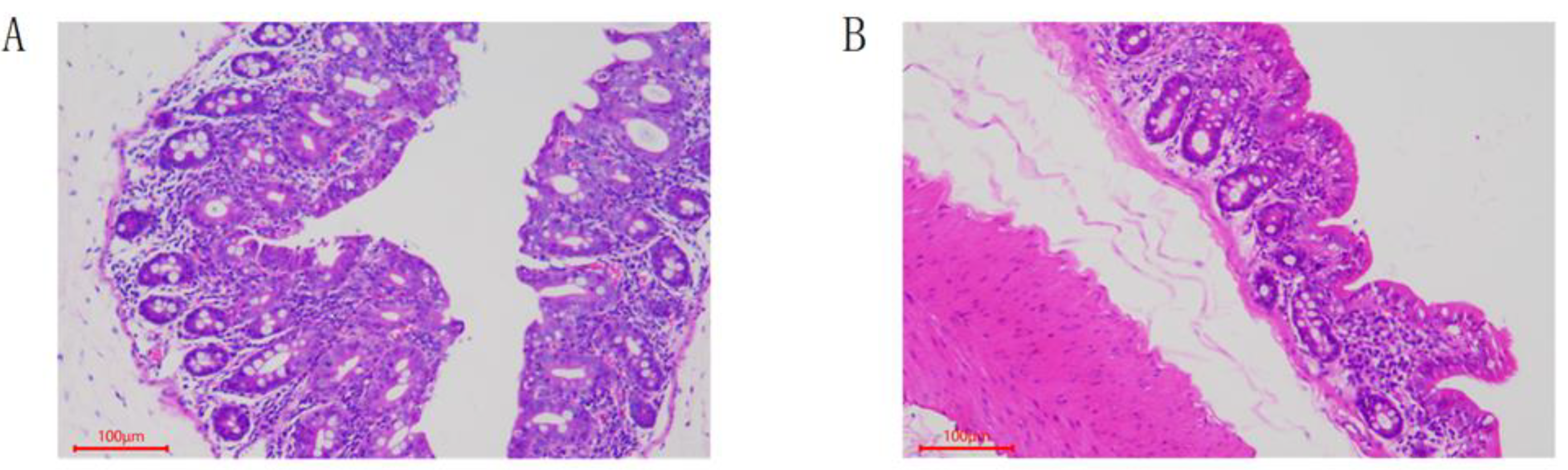
3.1. Pathological Features
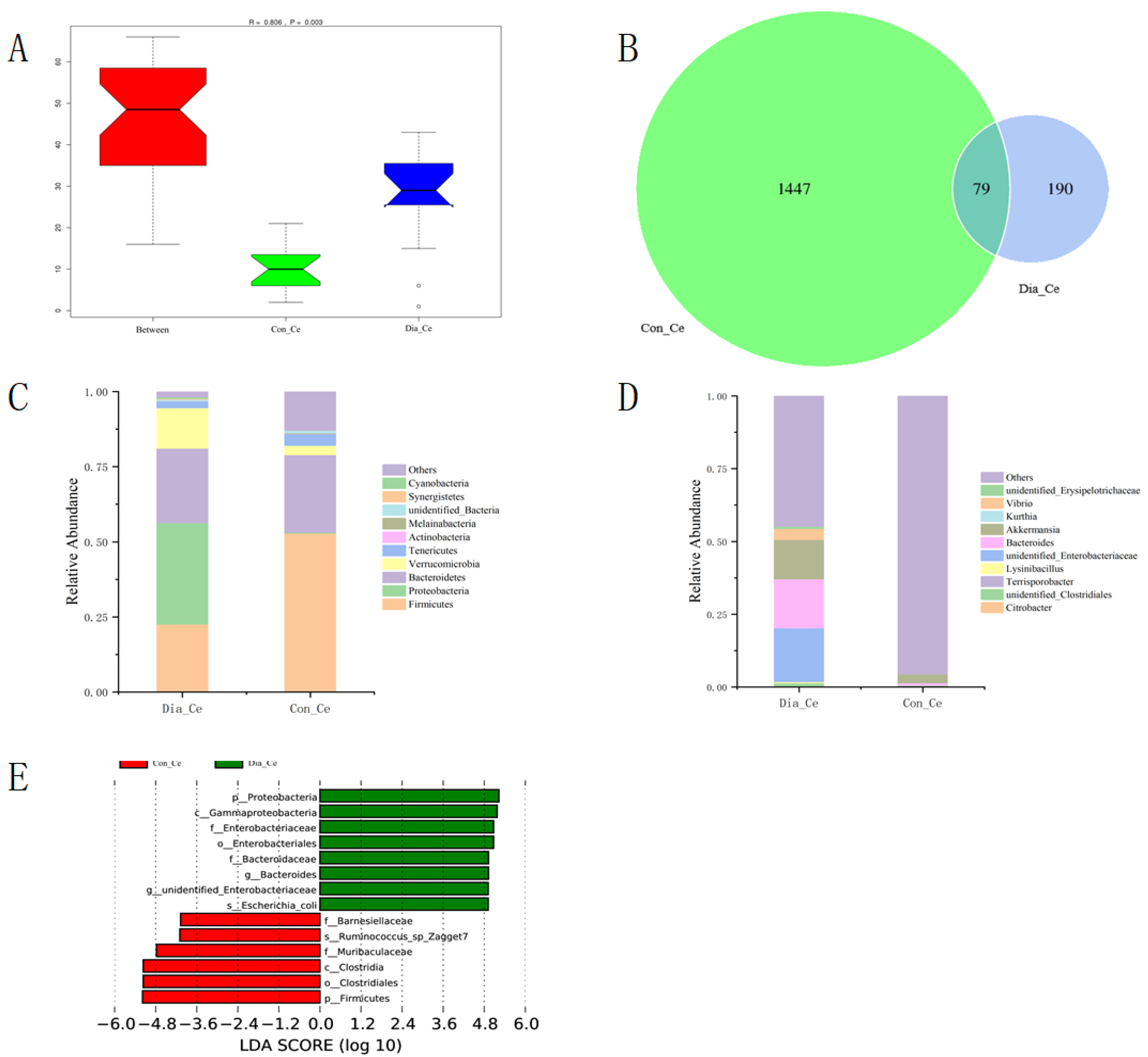
3.2. The Diversity of Fecal Microflora
3.3. The Transcriptome of Rabbits with Diarrhea
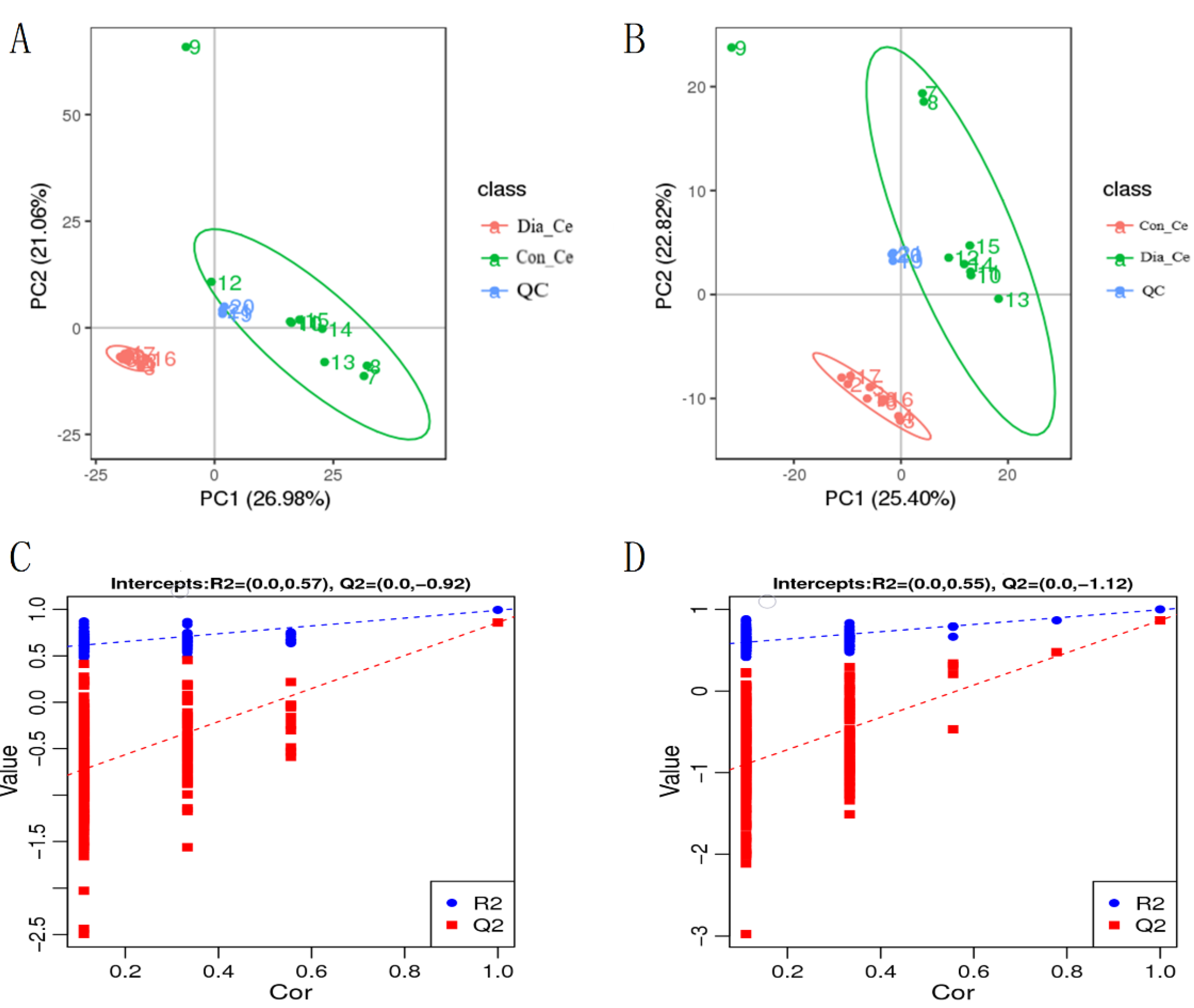
3.4. Analysis of Differential Metabolites
3.5. Correlation Analysis of DMs and the Cecum Bacteria and DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, Y.Q.; Ye, Q.; Zheng, W.Y. Research Status and Development of Antibiotics. World Notes Antibiot. 2019, 40, 295–301. [Google Scholar]
- Rosen, G.D. Applications of antibacterials in pig and poultry nutrition. Feed Comp. 1995, 15, 35–37. [Google Scholar]
- Danzeisen, J.L.; Kim, H.B.; Isaacson, R.E.; Tu, Z.J.; Johnson, T.J. Modulations of the Chicken Cecal Microbiome and Metagenome in Response to Anticoccidial and Growth Promoter Treatment. PLoS ONE 2011, 6, e27949. [Google Scholar] [CrossRef]
- Low, C.; Tan, L.; Ab Mutalib, N.-S.; Pusparajah, P.; Goh, B.-H.; Chan, K.-G.; Letchumanan, V.; Lee, L.-H. Unveiling the Impact of Antibiotics and Alternative Methods for Animal Husbandry: A Review. Antibiotics 2021, 10, 578. [Google Scholar] [CrossRef]
- Mingmongkolchai, S.; Panbangred, W. Bacillus probiotics: An alternative to antibiotics for livestock production. J. Appl. Microbiol. 2018, 124, 1334–1346. [Google Scholar] [CrossRef]
- Beltz, K.M.; Rosales, M.M.; Morales, E. Histological and ultrastructural findings in commercial bred rabbits exhibiting severe diarrhea. Scand. J. Lab. Anim. Sci. 2005, 32, 243–250. [Google Scholar]
- Prud′Hon, M.; Bel, L.J. Early weaning of young rabbits and reproduction in does. Ann. Zootech. 1968, 17, 23–30. [Google Scholar]
- Chen, Y.; Yan, S.; Zhu, C.; Yu, T.; Huang, W.; Huang, J.; Kong, Q.; Shi, J.; Chen, Z.; Liu, Q.; et al. Studying the Differences of Bacterial Metabolome and Microbiome in the Colon between Landrace and Meihua Piglets. Front. Microbiol. 2017, 8, 1812. [Google Scholar]
- Umesaki, Y.; Setoyama, H. Structure of the intestinal flora responsible for development of the gut immune system in a rodent model. Microbes and Infection 2000, 2, 1343–1351. [Google Scholar] [CrossRef]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Murphy, K.; Bloom, S.R. Gut hormones and the regulation of energy homeostasis. Nature 2006, 444, 854–859. [Google Scholar] [CrossRef]
- García, J.; Nicodemus, N.; Carabaño, R.; De Blass, J.C. Effect of inclusion of defatted grape seed meal in the diet on digestion and performance of growing rabbits. J. Anim. Sci. 2002, 80, 162–170. [Google Scholar] [CrossRef]
- Salisu, I.B.; Shahid, A.A.; Yaqoob, A.; Gul, A.; Rao, A.Q. Evaluation the effect of subchronic feeding of transgenic cotton line (CKC1) on the faecal microbiota of albino rabbits. J. Anim. Physiol. Anim. Nutr. 2021, 105, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Delgado, R.; Nicodemus, N.; Guaman, R.M.A.; Saste, J.; Menoyo, D.; Carabano, R.; Garcia, J. Effect of dietary soluble fibre and n-6/n-3 fatty acid ratio on growth performance and nitrogen and energy retention efficiency in growing rabbits. Anim. Feed Sci. Technol. 2018, 239, 44–54. [Google Scholar] [CrossRef]
- Crespo, R.; Alfonso, C.; del Barrio, A.S.; García-Ruiz, A.I.; Marco, M.; Nicodemus, N. Effect of feed restriction on performance, carcass yield and nitrogen and energy balance in growing rabbits. Livest. Sci. 2020, 241, 104278. [Google Scholar] [CrossRef]
- Zorn, A.M.; Wells, J.M. Vertebrate Endoderm Development and Organ Formation. Annu. Rev. Cell Dev. Biol. 2009, 25, 221–251. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Liang, G.; Guan, L.L. Regulation of rumen development in neonatal ruminants through microbial metagenomes and host transcriptomes. Genome Biol. 2019, 20, 172. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, L.; Li, P.; Li, X.; Zhou, H.; Wang, F.; Li, D.; Yin, Y.; Wu, G. Gene Expression Is Altered in Piglet Small Intestine by Weaning and Dietary Glutamine Supplementation. J. Nutr. 2008, 138, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.H.; Xiao, K.; Luan, Z.S.; Song, J. Early weaning increases intestinal permeability, alters expression of cytokine and tight junction proteins, and activates mitogen-activated protein kinases in pigs. J. Anim. Sci. 2013, 91, 1094–1101. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Li, Y.; Wang, J.; Elzo, M.; Shao, J.; Li, Y.; Xia, S.; Fan, H.; Jia, X.; Lai, S. Untargeted Metabolomics Reveals Intestinal Pathogenesis and Self-Repair in Rabbits Fed an Antibiotic-Free Diet. Animals 2021, 11, 1560. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Luo, Z.; Cao, C.; Sun, S.; Ma, Q.; Li, Z.; Shi, B.; Shan, A. Weaning Alters Intestinal Gene Expression Involved in Nutrient Metabolism by Shaping Gut Microbiota in Pigs Frontiers in Microbiology. Front. Microbiol. 2020, 11, 694. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, X.; Li, D.; Sun, J. Fecal Microbiota Transplantation Shows Marked Shifts in the Multi-Omic Profiles of Porcine Post-weaning Diarrhea. Front. Microbiol. 2021, 12, 619460. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bruns, M.A.; Tiedje, J.M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 1996, 62, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Christopher, Q.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplis, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Xie, D.; Lu, M.; Li, P.; Lv, H.; Yang, C.; Peng, Q.; Zhu, Y.; Guo, L.; Zhang, Y.; et al. Characterization of white tea metabolome: Comparison against green and black tea by a nontargeted metabolomics approach. Food Res. Int. 2017, 96, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Anne-Laure, B.; Strimmer, K. Partial least squares: A versatile tool for the analysis of high-dimensional genomic data. Brief. Bioinf. 2007, 8, 32–44. [Google Scholar]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef] [Green Version]
- Heischmann, S.; Quinn, K.; Cruickshank-Quinn, C.; Liang, L.-P.; Reisdorph, R.; Reisdorph, N.; Patel, M. Exploratory Metabolomics Profiling in the Kainic Acid Rat Model Reveals Depletion of 25-Hydroxyvitamin D3 during Epileptogenesis. Sci. Rep. 2016, 6, 31424. [Google Scholar] [CrossRef] [Green Version]
- Haspel, J.A.; Chettimada, S.; Shaik, R.S.; Chu, J.-H.; Raby, B.A.; Cernadas, M.; Carey, V.; Process, V.; Hunninghake, G.M.; Ifedigbo, E.; et al. Circadian rhythm reprogramming during lung inflammation. Nat. Commun. 2014, 5, 4753. [Google Scholar] [CrossRef]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Martinez, E.; Romano-Riquer, P.; Yanez-Marquez, E.; Longnecker, M.P.; Hernandez-Avila, M. Anogenital distance in human male and female newborns: A descriptive, cross-sectional study. Environ. Health 2004, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Abdelqader, A.; Al-Fataftah, A.-R.; Das, G. Effects of dietary Bacillus subtilis and inulin supplementation on performance, eggshell quality, intestinal morphology and microflora composition of laying hens in the late phase of production. Anim. Feed Sci. Technol. 2013, 179, 103–111. [Google Scholar] [CrossRef]
- Mountzouris, K.C.; Tsitrsikos, P.; Palamidi, I.; Arvaniti, A.; Mohnl, M.; Schatzmayr, G.; Fegeros, K. Effects of probiotic inclusion levels in broiler nutrition on growth performance, nutrient digestibility, plasma immunoglobulins, and cecal microflora composition. Poult. Sci. 2010, 89, 58–67. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, W.; Zhang, Z.; Liu, F.; Wang, J.; Yin, Y.; Wang, Z. Effects of Enterococcus faecalis on egg production, egg quality and caecal microbiota of hens during the late laying period. Arch. Anim. Nutr. 2019, 73, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Gulati, A.; Sartor, R.B. The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr. Opin. Gastroenterol. 2010, 26, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Dietert, R.R. The Microbiome in Early Life: Self-Completion and Microbiota Protection as Health Priorities. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2014, 101, 333–340. [Google Scholar] [CrossRef]
- Sleator, R.D. The human superorganism—Of microbes and men. Med. Hypotheses 2010, 74, 214–215. [Google Scholar] [CrossRef] [PubMed]
- Huffnagle, G.B. Chapter 40—Gastrointestinal Microbial Ecology with Perspectives on Health and Disease. In Physiology of the Gastrointestinal Tract; Elsevier Inc.: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Wexler, M.H. Bacteroides: The Good, the Bad, and the Nitty-Gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, C.; Ijaz, U.Z.; Gallagher, E.; Horton, F.; Ellis, R.J.; Jaiyeola, E.; Duparc, T.; Russell-Jones, D.; Hinton, P.; Cani, P.D.; et al. Fecal Enterobacteriales enrichment is associated with increased invivo intestinal permeability in humans. Physiol. Rep. 2018, 6, e13649. [Google Scholar] [CrossRef]
- Saemann, M.D.; Bohmig, G.A.; Osterreicher, C.H.; Burtscher, H.; Parolini, O.; Diakos, C.; Stockl, J.; Horl, W.H.; Zlabinger, G.J. Anti-inflammatory effects of sodium butyrate on human monocytes: Potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB J. 2000, 14, 2380–2382. [Google Scholar] [CrossRef]
- Cavaglieri, C.R.; Nishiyama, A.; Fernandes, L.C.; Curi, R.; Miles, E.A.; Calder, P.C. Differential effects of short-chain fatty acids on proliferation and production of pro- and anti-inflammatory cytokines by cultured lymphocytes. Life Sci. 2003, 73, 1683–1690. [Google Scholar] [CrossRef]
- Zwicker, B.L.; Agellon, L.B. Transport and biological activities of bile acids. Int. J. Biochem. Cell Biol. 2013, 45, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Praslickova, D.; Torchia, E.C.; Sugiyama, M.G.; Magrane, E.J.; Zwicker, B.L.; Kolodzieyski, L.; Agellon, L.B. The Ileal Lipid Binding Protein Is Required for Efficient Absorption and Transport of Bile Acids in the Distal Portion of the Murine Small Intestine. PLoS ONE 2012, 7, e50810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, K.; Arai, S.; Itoh, H.; Adachi, S.; Hayashida, M.; Nakase, H.; Ikemoto, M. CD68 on rat macrophages binds tightly to S100A8 and S100A9 and helps to regulate the cells’ immune functions. J. Leukoc. Biol. 2016, 100, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- Vereecke, L.; Beyaert, R.; van Loo, G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009, 30, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-Y.; Zhu, M.; Zhu, B.; Wang, Z.-Q. Cholera Toxin Suppresses Expression of Ubiquitin Editing Enzyme A20 and Enhances Transcytosis. Cell. Physiol. Biochem. 2013, 31, 495–504. [Google Scholar] [CrossRef]
- Vereecke, L.; Vieira-Silva, S.; Billiet, T.; Van Es, J.H.; Mc Guire, C.; Slowicka, K.; Sze, M.; Born, M.V.D.; De Hertogh, G.; Clevers, H.; et al. A20 controls intestinal homeostasis through cell-specific activities. Nat. Commun. 2014, 5, 5103. [Google Scholar] [CrossRef] [Green Version]
- Vereecke, L.; Sze, M.; Mc Guire, C.; Rogiers, B.; Chu, Y.; Schmidt-Supprian, M.; Pasparakis, M.; Beyaert, R.; van Loo, G. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor–induced toxicity and experimental colitis. J. Exp. Med. 2010, 207, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Kolodziej, L.E.; Lodolce, J.P.; Chang, J.E.; Schneider, J.R.; Grimm, W.A.; Bartulis, S.J.; Zhu, X.; Messer, J.S.; Murphy, S.F.; Reddy, N.; et al. TNFAIP3 Maintains Intestinal Barrier Function and Supports Epithelial Cell Tight Junctions. PLoS ONE 2011, 6, e26352. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, T.; Jinnin, M.; Yamane, K.; Honda, N.; Kajihara, I.; Makino, T.; Masuguchi, S.; Fukushima, S.; Okamoto, Y.; Hasegawa, M.; et al. Impaired IL-17 Signaling Pathway Contributes to the Increased Collagen Expression in Scleroderma Fibroblasts. J. Immunol. 2012, 188, 3573–3583. [Google Scholar] [CrossRef] [Green Version]
- Iwakura, Y.; Ishigame, H. The IL-23/IL-17 axis in inflammation. J. Clin. Investig. 2006, 116, 1218. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.-N.; Kang, M.-J.; Lee, S.-J.; Kim, J.-I. Ameliorative effect of myricetin on insulin resistance in mice fed a high-fat, high-sucrose diet. Nutr. Res. Pr. 2014, 8, 544–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Zhang, Z.-F.; Dai, X.-Q.; Li, Y. Myricetin Protects Against Cytokine-Induced Cell Death in RIN-m5f β Cells. J. Med. Food 2012, 15, 733–740. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Gu, D.; Huang, Y.; Ayupbek, A.; Yang, Y.; Aisa, H.A.; Ito, Y. Separation and Purification of Phenolic Acids and Myricetin from Black Currant by High-Speed Countercurrent Chromatography. J. Liq. Chromatogr. Relat. Technol. 2009, 32, 3077–3088. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ma, S.; Ning, M.; Yang, W.; Ye, Y.; Zhang, L.; Shen, J.; Leng, Y. TGR5 agonist ameliorates insulin resistance in skeletal muscles and improves glucose homeostasis in diabetic mice. Metabolism 2019, 99, 45–56. [Google Scholar] [CrossRef]
- Lazarevi, S.; Danic, M.; Golocorbin-Kon, S.; Al-Salami, H.; Mikov, M. Semisynthetic bile acids: A new therapeutic option for metabolic syndrome. Pharmacol. Res. 2019, 146, 104333. [Google Scholar] [CrossRef]
- Song, P.; Rockwell, C.E.; Cui, J.Y.; Klaassen, C.D. Individual bile acids have differential effects on bile acid signaling in mice. Toxicol. Appl. Pharmacol. 2015, 283, 57–64. [Google Scholar] [CrossRef]
- STanimirov, B.; Stankov, K.; Mikov, M. Bile acid signaling through farnesoid X and TGR5 receptors in hepatobiliary and intestinal diseases. Hepatobil. Pancreat. Dis. Int. 2015, 14, 18–33. [Google Scholar] [CrossRef]
- Münch, A.; Ström, M.; Söderholm, J.D. Dihydroxy bile acids increase mucosal permeability and bacterial uptake in human colon biopsies. Scand. J. Gastroenterol. 2007, 42, 1167–1174. [Google Scholar] [CrossRef]
- Ward, J.B.J.; Lajczak, N.K.; Kelly, O.B.; O’Dwyer, A.M.; Giddam, A.K.; Gabhann, J.N.; Franco, P.; Tambuwala, M.M.; Jefferies, C.A.; Keely, S.; et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am. J. Physiol. Liver Physiol. 2017, 312, G550–G558. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, A.A.; Ramachandran, D.; Chaudhari, S.N.; Powell, C.E.; Li, W.; McCurry, M.D.; Banks, A.S.; Devlin, A.S. A Gut-Restricted Lithocholic Acid Analog as an Inhibitor of Gut Bacterial Bile Salt Hydrolases. ACS Chem. Biol. 2021, 16, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Blacher, E.; Elinav, E. Microbiome, metabolites and host immunity. Curr. Opin. Microbiol. 2017, 35, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Jin, U.-H.; Allred, C.D.; Jayaraman, A.; Chapkin, R.S.; Safe, S. Aryl Hydrocarbon Receptor Activity of Tryptophan Metabolites in Young Adult Mouse Colonocytes. Drug Metab. Dispos. 2015, 43, 1536–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Togo, A.; Durand, G.; Khelaifia, S.; Armstrong, N.; Robert, C.; Cadoret, F.; Di Pinto, F.; Delerce, J.; Levasseur, A.; Raoult, D.; et al. Fournierella massiliensis gen. nov., sp. nov., a new human-associated member of the family Ruminococcaceae. Int. J. Syst. Evol. Microbiol. 2017, 67, 1393–1399. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Observed | Chao1 | Shannon | Simpson | Coverage (%) |
|---|---|---|---|---|---|
| Dia_Ce | 75.50 | 112.87 | 3.62 | 0.78 | 96.43 |
| Con_Ce | 336.33 | 1304.05 | 6.59 | 0.95 | 71.68 |
| SEM | 98.794 | 619.389 | 0.802 | 0.059 | 11,246 |
| p-value | 0.025 | 0.083 | 0.004 | 0.0163 | 0.052 |
| Group | Clostridiales | Enterobacteriales | Bacillales | Bacteroidales | Verrucomicrobiales | Erysipelotrichales |
|---|---|---|---|---|---|---|
| Dia_Ce | 0.205404 | 0.264739 | 0.005669 | 0.243197 | 0.134921 | 0.013228 |
| Con_Ce | 0.493008 | 0 | 0 | 0.256992 | 0.03099 | 0.00737 |
| SEM | 0.087 | 0.149 | 0.004 | 0.183 | 0.092 | 0.005 |
| p-value | 0.009 | 0.106 | 0.164 | 0.899 | 0.283 | 0.273 |
| Sample | Total Reads | Total Map | Unique Map | Multi Map | Positive Map | Negative Map |
|---|---|---|---|---|---|---|
| Dia_Ce_1 | 47079708 | 41,829,313 (88.85%) | 36,717,519 (77.99%) | 5,111,794 (10.86%) | 18,315,048 (38.9%) | 18,402,471 (39.09%) |
| Dia_Ce_2 | 40705516 | 36,216,676 (88.97%) | 33,688,313 (82.76%) | 2,528,363 (6.21%) | 16,836,322 (41.36%) | 16,851,991 (41.4%) |
| Dia_Ce_3 | 44406510 | 38,983,466 (87.79%) | 36,626,529 (82.48%) | 2,356,937 (5.31%) | 18,271,015 (41.14%) | 18,355,514 (41.34%) |
| Con_Ce_1 | 43584230 | 38,754,099 (88.92%) | 34,649,374 (79.5%) | 4,104,725 (9.42%) | 17,294,670 (39.68%) | 17,354,704 (39.82%) |
| Con_Ce_2 | 40285156 | 34,846,494 (86.5%) | 31,066,590 (77.12%) | 3,779,904 (9.38%) | 15,488,047 (38.45%) | 15,578,543 (38.67%) |
| Con_Ce_3 | 46651768 | 39,602,947 (84.89%) | 37,114,380 (79.56%) | 2,488,567 (5.33%) | 18,502,785 (39.66%) | 18,611,595 (39.89%) |
| Compared Samples | Num. of Total Ident. | Num. of Total Sig. | Num. of Sig. Up | Num. of Sig. Down |
|---|---|---|---|---|
| Dia_Ce. vs. Con_Ce. pos | 1251 | 485 | 307 | 178 |
| Dia_Ce. vs. Con_Ce. neg | 493 | 167 | 102 | 65 |
| Map ID | Map Title | p-Value | N | Meta IDs | |
|---|---|---|---|---|---|
| ESI- | map04976 | Bile secretion | 0.07369 | 105 | Salicylic acid, Reduced glutathione, Thromboxane B2, Lithocholic Acid, Chenodeoxycholic Acid, Deoxycholic acid |
| map00520 | Amino sugar and nucleotide sugar metabolism | 0.098581 | 105 | L-Fucose, N-Acetylneuraminic acid, N-Acetyl-α-D-glucosamine 1-phosphate | |
| map00380 | Tryptophan metabolism | 0.244399 | 105 | Quinolinic acid, Picolinic acid | |
| map04152 | AMPK signaling pathway | 0.32381 | 105 | NAD+ | |
| ESI+ | map01523 | Antifolate resistance | 0.072914 | 182 | Folic acid, Guanosine monophosphate, Adenosine 5′-monophosphate, 7,8-Dihydrofolate |
| map04742 | Taste transduction | 0.072914 | 182 | Serotonin, Guanosine monophosphate, D-Phenylalanine, Adenosine 5′-monophosphate | |
| map00592 | alpha-Linolenic acid metabolism | 0.146621 | 182 | Jasmonic acid, Traumatic acid |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Zhao, K.; Kang, Z.; Wang, M.; Chen, Y.; Fan, H.; Xia, S.; Lai, S. The Multi-Omics Analysis Revealed a Metabolic Regulatory System of Cecum in Rabbit with Diarrhea. Animals 2022, 12, 1194. https://doi.org/10.3390/ani12091194
Wang J, Zhao K, Kang Z, Wang M, Chen Y, Fan H, Xia S, Lai S. The Multi-Omics Analysis Revealed a Metabolic Regulatory System of Cecum in Rabbit with Diarrhea. Animals. 2022; 12(9):1194. https://doi.org/10.3390/ani12091194
Chicago/Turabian StyleWang, Jie, Kaisen Zhao, Zhe Kang, Meigui Wang, Yang Chen, Huimei Fan, Siqi Xia, and Songjia Lai. 2022. "The Multi-Omics Analysis Revealed a Metabolic Regulatory System of Cecum in Rabbit with Diarrhea" Animals 12, no. 9: 1194. https://doi.org/10.3390/ani12091194
APA StyleWang, J., Zhao, K., Kang, Z., Wang, M., Chen, Y., Fan, H., Xia, S., & Lai, S. (2022). The Multi-Omics Analysis Revealed a Metabolic Regulatory System of Cecum in Rabbit with Diarrhea. Animals, 12(9), 1194. https://doi.org/10.3390/ani12091194