
Gut Microbiome Profiling of the Endangered Southern Greater Glider (Petauroides volans) after the 2019–2020 Australian Megafire



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
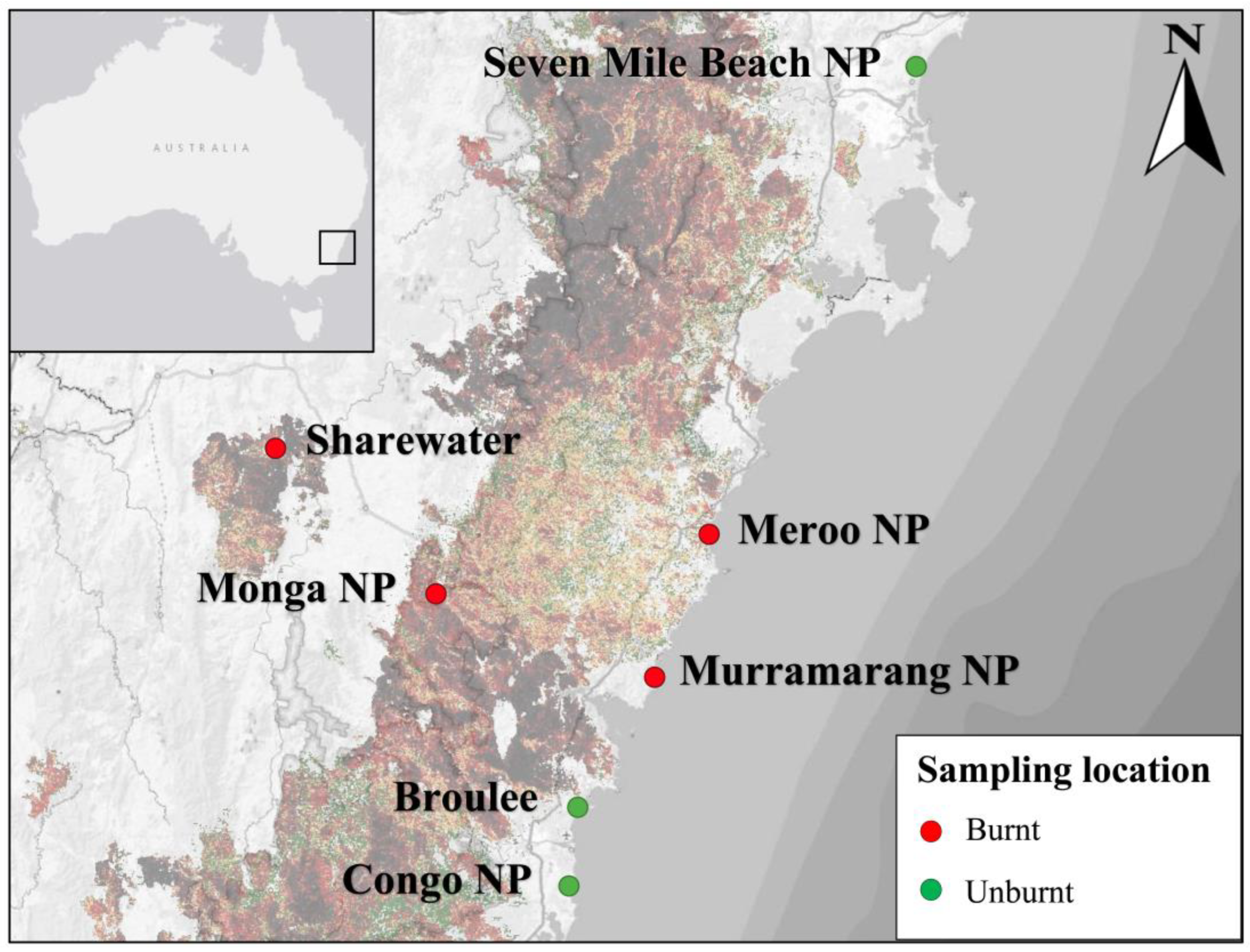
2.1. Study Area
2.2. Sample Collection
2.3. DNA Extraction and 16S Microbial Diversity Profiling
2.4. Microbial Diversity Analyses
2.5. Taxonomic Composition Profiling
2.6. Prediction of Functional Profiles of Microbial Communities
2.7. Differential Abundance Analysis
2.8. Data Availability
3. Results
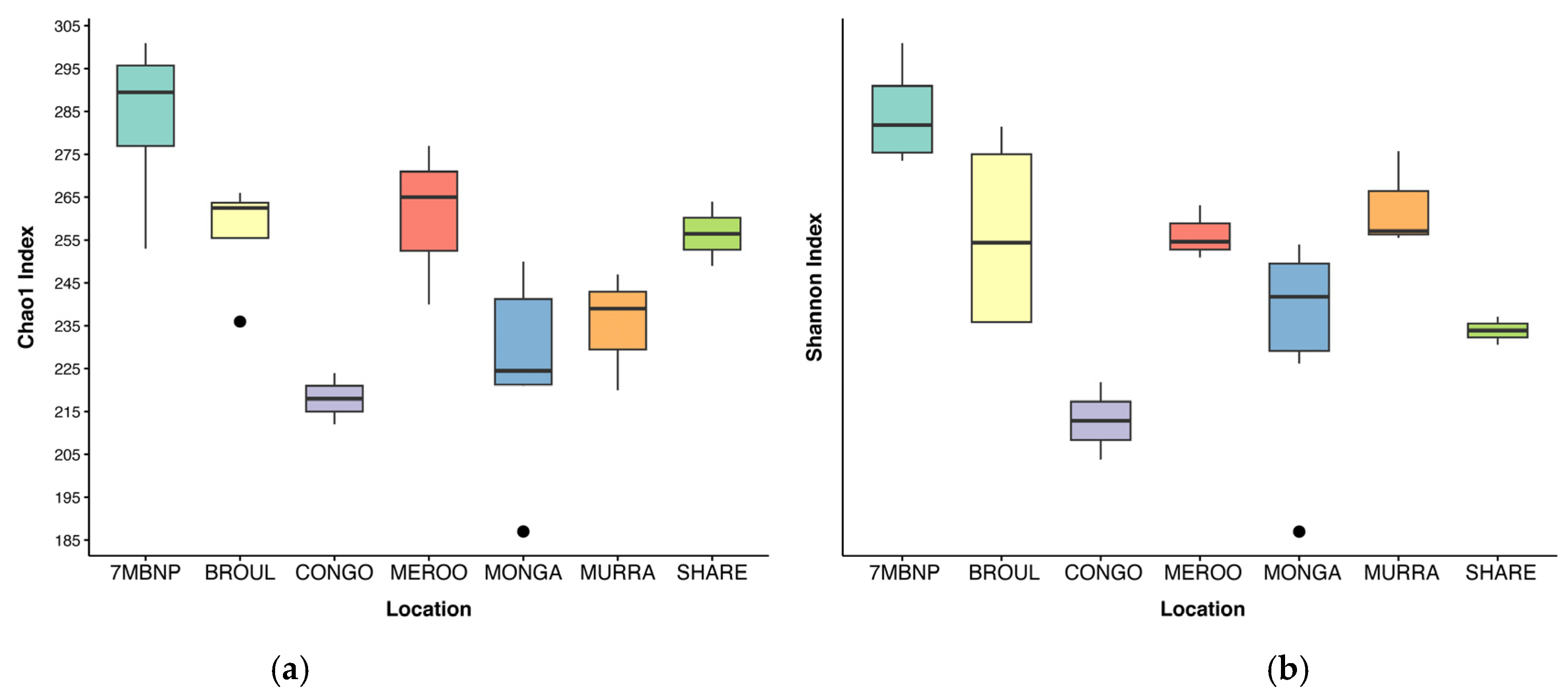
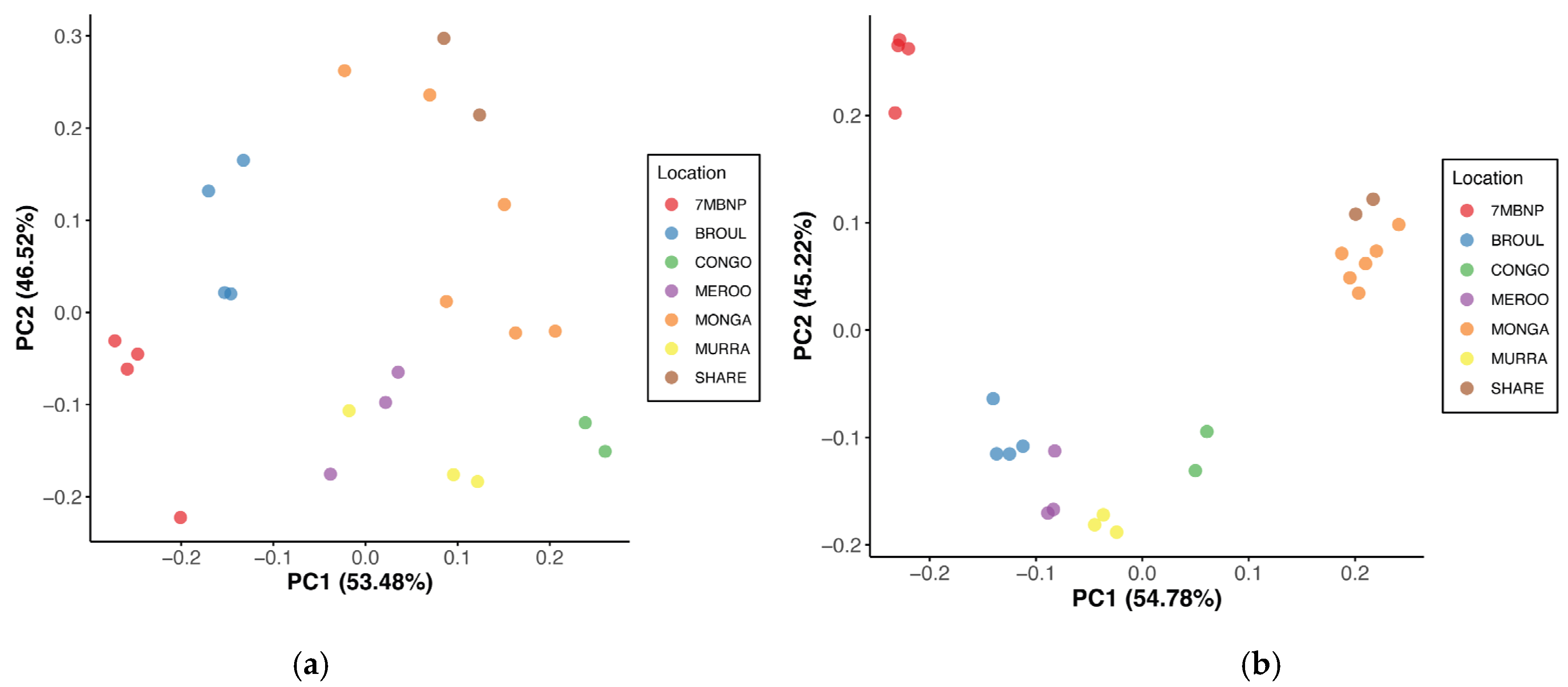
3.1. Microbial Diversity
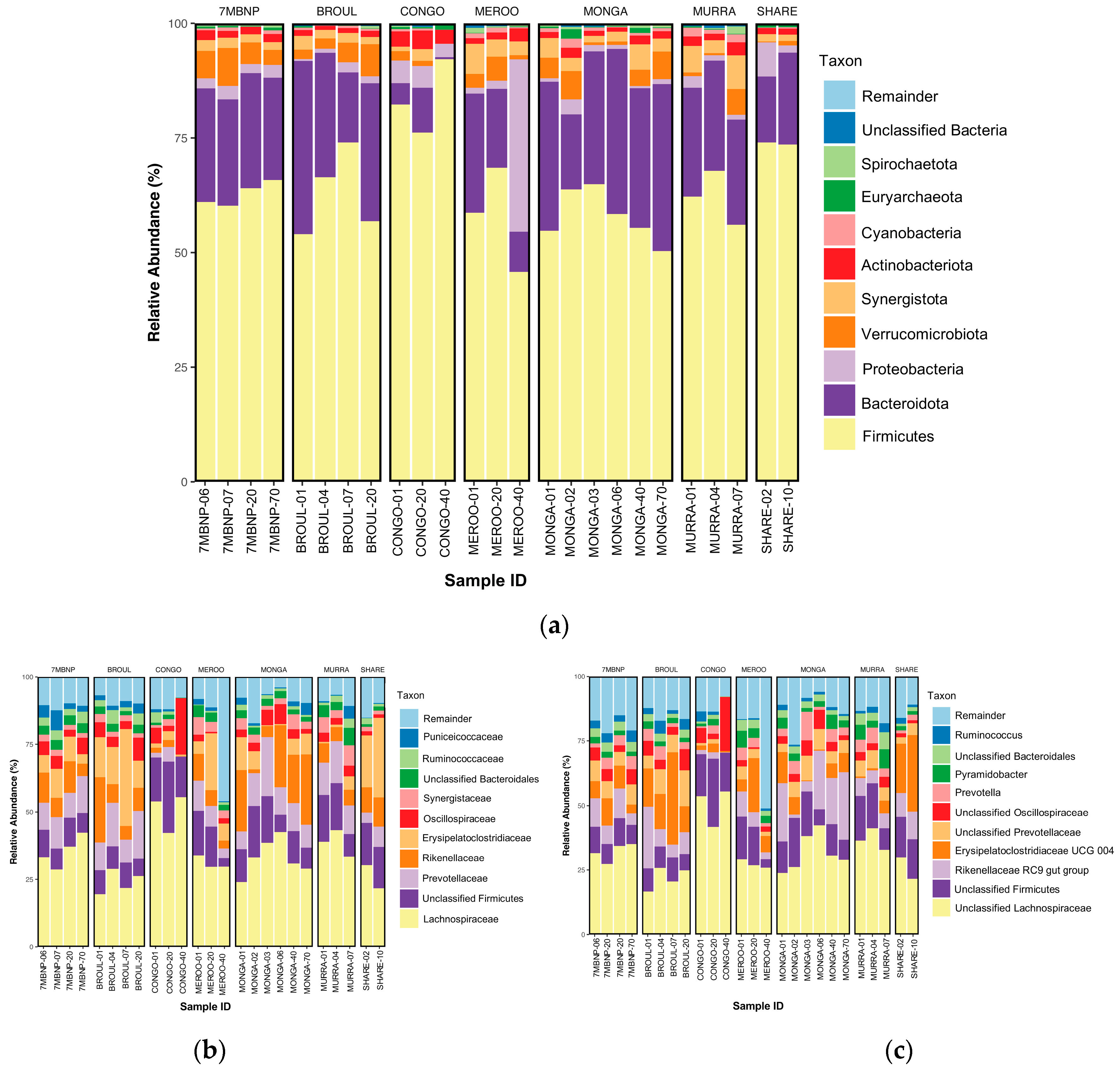
3.2. Taxonomic Composition
3.2.1. Phylum Level
3.2.2. Family Level
3.2.3. Genus Level
3.3. Functional Profiling of Microbial Communities
3.4. Differential Abundance Analysis
4. Discussion
4.1. Southern Greater Glider Gut Microbiomes Exhibit Varied Microbial Diversity across the Landscape
4.2. Southern Greater Glider Gut Microbiomes Are Taxonomically Diverse
4.3. Gut Microbial Community Functional Profiles Provide Insights into Greater Glider Metabolism, Diet and Health
4.4. Wildfire Affects the Presence and Abundance of Arboreal Marsupial Gut Microbiota
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Location | Dominant Vegetation Classes | Reported Eucalyptus Species | Monthly Mean Temperature Range for 2021 (°C) | Annual Mean Rainfall for 2021 (mm) | 2019–2020 Fires Burn Status | Greater Glider Effective Population Size |
|---|---|---|---|---|---|---|
| Seven Mile Beach National Park, Gerroa | South Coast Sands Dry Sclerophyll Forests | E. botryoides E. pilularis | 20.9 | 1580.6 | Unburnt | 45.1 |
| South Broulee Beach, Broulee | South Coast Sands Dry Sclerophyll Forests | E. botryoides E. pilularis | 20.1 | 1189.4 | Unburnt | 87.7 |
| Eurobodalla National Park, Congo | Southern Lowland Wet Sclerophyll Forests | E. pilularis E. botyroides E. scias E. agglomerata E. paniculata | 20.9 | 1144.9 | Unburnt | 6.2 |
| Meroo National Park, Meroo | Southern Lowland Wet Sclerophyll Forests; Southeast Dry Sclerophyll Forests | E. pilularis E. botyroides E. scias E. agglomerata E. paniculata E. agglomerata E. globoidea E. sieberi E. consididenia | 22.6 | 969.2 | Burnt | 8.2 |
| Monga National Park, Monga | Southern Escarpment Wet Sclerophyll Forests | E. cypellocarpa E. fastigata E. obliqua E. viminalis | 18.4 | 1473.0 | Burnt | 160.8 |
| Murramarang National Park, Murramurang | Southeast Dry Sclerophyll Forests | E. agglomerata E. paniculata E. agglomerata E. globoidea E. sieberi E. consididenia | 22.0 | 1092.4 | Burnt | 7.8 |
| Sharewater, Tallaganda | Southern Escarpment Wet Sclerophyll Forests; Southeast Dry Sclerophyll Forests | E. cypellocarpa E. fastigata E. obliqua E. viminalis E. agglomerata E. paniculata E. agglomerata E. globoidea E. sieberi E. blaxlandii E. dives E. smithii | 18.4 | 1261.4 | Burnt | - |
| ASV | Taxonomic Classification | W | Mean Relative Abundance (% ± SEM) | |
|---|---|---|---|---|
| 27a92e9d957ed8ff16c0dfac8033bdf5 | d__Bacteria; p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae | 1091 | Sharewater | 2.11 ± 0.07 |
| Monga NP | 1.71 ± 0.18 | |||
| 24c0bed7fe8e9346c4a624644cf979e8 | d__Bacteria; p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae | 1091 | Murramarang NP | 2.59 ± 0.99 |
| Meroo NP | 0.97 ± 0.37 | |||
| d1d1a5a618360d5a64a3e9fe0a39e394 | d__Bacteria; p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae | 1090 | Broulee | 0.81 ± 0.22 |
| Eurobodalla NP | 0.80 ± 0.12 | |||
| Seven Mile Beach NP | 0.60 ± 0.16 | |||
| bc2343861ddbca17c55ee68a21fe8e3a | d__Bacteria; p__Firmicutes | 1090 | Seven Mile Beach NP | 3.52 ± 0.20 |
| 28b2ac3cac0b46d8b26a31f1ab59c922 | d__Bacteria; p__Actinobacteriota; c__Coriobacteriia; o__Coriobacteriales; f__Eggerthellaceae; g__Enterorhabdus; s__uncultured_bacterium | 1084 | Sharewater | 0.41 ± 0.10 |
| Monga NP | 0.38 ± 0.09 | |||
| Seven Mile Beach NP | 0.24 ± 0.06 | |||
| c001095135fb918b7c1715ddecc36fa6 | d__Bacteria; p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae | 1082 | Monga NP | 0.51 ± 0.14 |
| Seven Mile Beach NP | 0.28 ± 0.08 | |||
| 8253eb9289121ce88046e7ebb4b642e5 | d__Bacteria; p__Actinobacteriota; c__Coriobacteriia; o__Coriobacteriales; f__Eggerthellaceae; g__Enterorhabdus; s__uncultured_bacterium | 1071 | Eurobodalla NP | 0.99 ± 0.31 |
| Murramarang NP | 0.64 ± 0.15 | |||
| Meroo NP | 0.29 ± 0.09 | |||
| Seven Mile Beach NP | 0.21 ± 0.03 | |||
| Broulee | 0.17 ± 0.04 | |||
| f73e366c345a0d82a70413c5aec880b6 | d__Bacteria; p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae | 1009 | Eurobodalla NP | 0.74 ± 0.04 |
| Monga NP | 0.89 ± 0.44 | |||
| Meroo NP | 0.27 ± 0.07 | |||
| Murramarang NP | 0.07 ± 0.03 | |||
| 1bc20c352c5297c1e260d58aabeeb750 | d__Bacteria; p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae | 1033 | Eurobodalla NP | 5.04 ± 2.00 |
| Sharewater | 2.30 ± 0.35 | |||
| Monga NP | 1.57 ± 0.63 | |||
| Broulee | 0.51 ± 0.18 | |||
| Meroo NP | 0.34 ± 0.04 | |||
| Murramarang NP | 0.33 ± 0.14 | |||
| e13ba7436b454d9525ce3e631b789355 | d__Bacteria; p__Verrucomicrobiota; c__Verrucomicrobiae; o__Opitutales; f__Puniceicoccaceae; g__Cerasicoccus; s__uncultured_bacterium | 1065 | Seven Mile Beach NP | 4.02 ± 1.72 |
| bd537de474dbb1b14c5cc52cc017a309 | d__Bacteria; p__Bacteroidota; c__Bacteroidia; o__Bacteroidales | 1046 | Monga NP | 0.39 ± 0.15 |
| Murramarang NP | 0.34 ± 0.06 | |||
| Broulee | 0.30 ± 0.05 | |||
| Eurobodalla NP | 0.22 ± 0.17 | |||
| Sharewater | 0.11 ± 0.03 | |||
| 50f8d53a33780a64348ec66e38a33cdf | d__Bacteria; p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae | 1026 | Eurobodalla NP | 1.95 ± 1.58 |
| Monga NP | 0.33 ± 0.11 | |||
| Broulee | 0.05 ± 0.01 | |||
| Murramarang NP | 0.03 ± 0.03 | |||
| 0786c15611bf814f2d41cf851a71b2b6 | d__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Burkholderiales | 1048 | Seven Mile Beach NP | 1.35 ± 0.10 |
| b47ecf8a5559f8e193a4ca3cdbf074b6 | d__Bacteria; p__Firmicutes; c__Clostridia; o__Oscillospirales; f__Ruminococcaceae; g__Ruminococcus | 1049 | Broulee | 1.67 ± 0.79 |
| 021fe8edae51250b230154489e2dfb4c | d__Bacteria; p__Bacteroidota; c__Bacteroidia; o__Bacteroidales; f__Prevotellaceae | 1026 | Seven Mile Beach NP | 1.11 ± 0.28 |
| a75e2f333dcb0988db971ba7d81e950b | d__Bacteria; p__Bacteroidota; c__Bacteroidia; o__Bacteroidales; f__Prevotellaceae | 1013 | Murramarang NP | 0.38 ± 0.28 |
| Meroo NP | 0.19 ± 0.05 | |||
| Broulee | 0.10 ± 0.01 | |||
| 41ef40105853acf4d6452e5e3c3859da | d__Bacteria; p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae | 998 | Murramarang NP | 1.62 ± 0.24 |
| Meroo NP | 1.53 ± 0.36 | |||
| Seven Mile Beach NP | 0.43 ± 0.14 | |||
| Broulee | 0.39 ± 0.08 | |||
| Eurobodalla NP | 0.22 ± 0.03 | |||
| Sharewater | 0.12 ± 0.12 | |||
| 0656d5259d674955e7a3e2d1d511bc10 | d__Bacteria; p__Verrucomicrobiota; c__Verrucomicrobiae; o__Opitutales; f__Puniceicoccaceae; g__Cerasicoccus; s__uncultured_bacterium | 1002 | Seven Mile Beach NP | 0.67 ± 0.36 |
| 820a74cb34678c22bddf0be11d75e893 | d__Bacteria; p__Firmicutes; c__Clostridia; o__Lachnospirales; f__Lachnospiraceae | 1000 | Sharewater | 0.49 ± 0.19 |
| Broulee | 0.42 ± 0.11 | |||
| Murramarang NP | 0.31 ± 0.07 | |||
| Monga NP | 0.02 ± 0.02 | |||
| 9f5d77c743e4e8fd92851d3fe40b323e | d__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Burkholderiales | 994 | Broulee | 0.21 ± 0.04 |
| Eurobodalla NP | 0.12 ± 0.02 | |||
| 01cf9a65286e6b93cc6c83aef2e7f19e | d__Bacteria; p__Bacteroidota; c__Bacteroidia; o__Bacteroidales | 988 | Seven Mile Beach NP | 1.05 ± 0.24 |
| d9a15c071f0df2e405f60ebbd8d5cb61 | d__Bacteria; p__Bacteroidota; c__Bacteroidia; o__Bacteroidales | 982 | Murramarang NP | 0.41 ± 0.13 |
| Meroo NP | 0.39 ± 0.03 | |||
| Broulee | 0.21 ± 0.05 | |||
| Eurobodalla NP | 0.07 ± 0.07 | |||
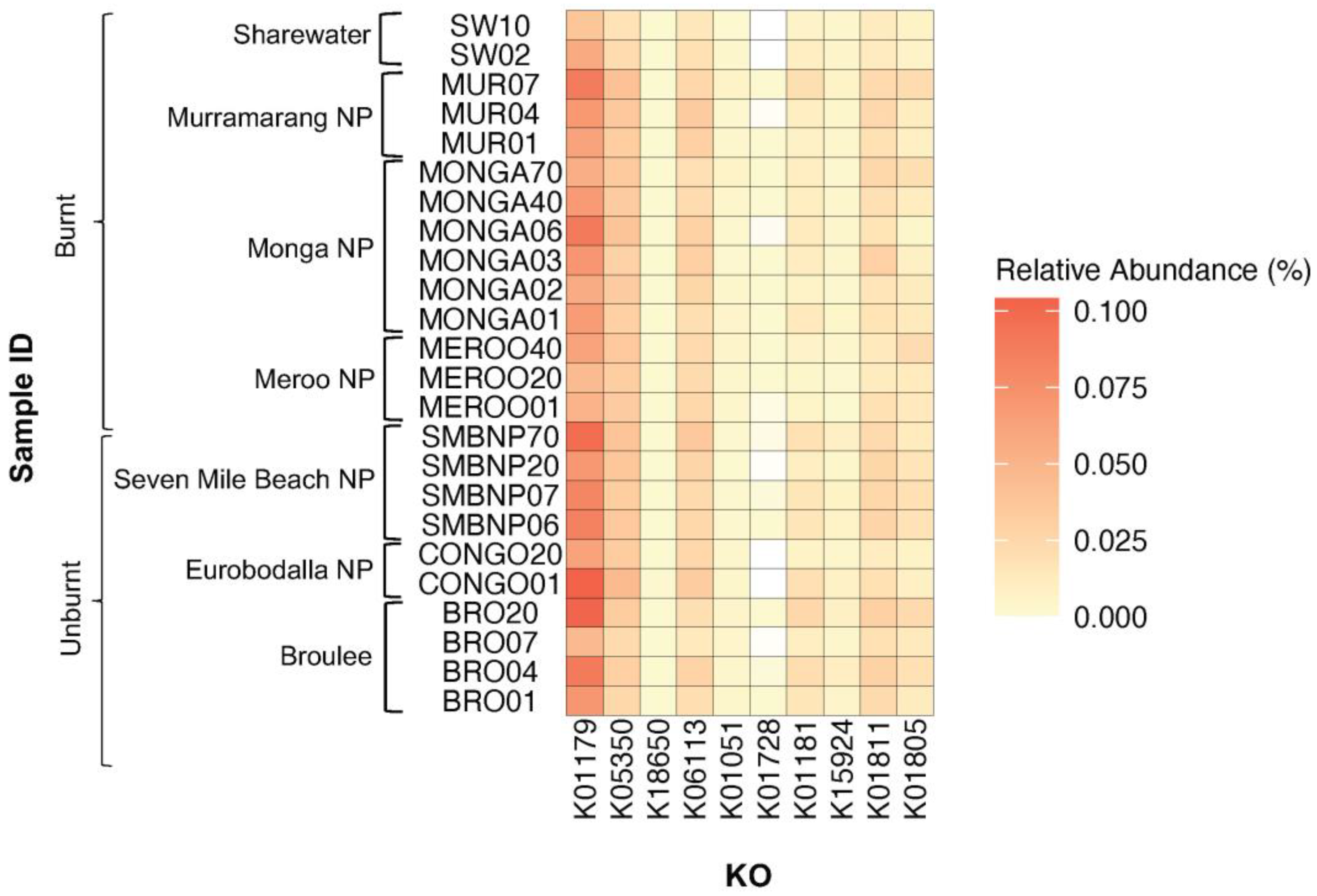
| Fibre Degraded | KEGG Orthologue | Enzyme | Mean Relative Abundance (% ± SEM) | p Value | |
|---|---|---|---|---|---|
| Cellulose | K01179 | Endoglucanase | Broulee | 0.077% ± 0.012% | 0.10 |
| Eurobodalla NP | 0.094% ± 0.016% | ||||
| Meroo NP | 0.053% ± 0.0050% | ||||
| Monga NP | 0.068% ± 0.0053% | ||||
| Murramarang NP | 0.072% ± 0.0078% | ||||
| Seven Mile Beach NP | 0.083% ± 0.0059% | ||||
| Sharewater | 0.048% ± 0.0096% | ||||
| K05350 | Beta-glucosidase | Broulee | 0.028% ± 0.0026% | 0.07 | |
| Eurobodalla NP | 0.037% ± 0.0041% | ||||
| Meroo NP | 0.033% ± 0.0010% | ||||
| Monga NP | 0.033% ± 0.0015% | ||||
| Murramarang NP | 0.036% ± 0.0023% | ||||
| Seven Mile Beach NP | 0.036% ± 0.0016% | ||||
| Sharewater | 0.020% ± 0.0022% | ||||
| Pectin | K18650 | Exo-poly-alphagalacturonosidase | Broulee | 0.00048% ± 0.000046% | 0.03 * |
| Eurobodalla NP | 0.00093% ± 0.00029% | ||||
| Meroo NP | 0.00032% ± 0.000027% | ||||
| Monga NP | 0.00060% ± 0.00010% | ||||
| Murramarang NP | 0.00078% ± 0.000024% | ||||
| Seven Mile Beach NP | 0.00085% ± 0.000039% | ||||
| Sharewater | 0.00040% ± 0.0000052% | ||||
| K06113 | Arabinan endo-1,5-alpha-Larabinosidase | Broulee | 0.021% ± 0.0029% | 0.18 | |
| Eurobodalla NP | 0.025% ± 0.0048% | ||||
| Meroo NP | 0.024% ± 0.00057% | ||||
| Monga NP | 0.024% ± 0.0020% | ||||
| Murramarang NP | 0.030% ± 0.0025% | ||||
| Seven Mile Beach NP | 0.027% ± 0.0029% | ||||
| Sharewater | 0.017% ± 0.0017% | ||||
| K01051 | Pectinesterase | Broulee | 0.0040% ± 0.00053% | 0.10 | |
| Eurobodalla NP | 0.0029% ± 0.00081% | ||||
| Meroo NP | 0.0034% ± 0.00097% | ||||
| Monga NP | 0.0035% ± 0.00083% | ||||
| Murramarang NP | 0.0033% ± 0.0017% | ||||
| Seven Mile Beach NP | 0.0013% ± 0.000091% | ||||
| Sharewater | 0.0019% ± 0.00071% | ||||
| K01728 | Pectate lyase | Broulee | 0.00020% ± 0.000088% | 0.25 | |
| Eurobodalla NP | 0.00011% ± 0.00011% | ||||
| Meroo NP | 0.00016% ± 0.000064% | ||||
| Monga NP | 0.00024% ± 0.000098% | ||||
| Murramarang NP | 0.00017% ± 0.000078% | ||||
| Seven Mile Beach NP | 0.000075% ± 0.000027% | ||||
| Sharewater | - | ||||
| Xylan | K01181 | Endo-1,4-beta-xylanase | Broulee | 0.018% ± 0.0031% | 0.04 * |
| Eurobodalla NP | 0.015% ± 0.0039% | ||||
| Meroo NP | 0.0053% ± 0.00049% | ||||
| Monga NP | 0.011% ± 0.0011% | ||||
| Murramarang NP | 0.012% ± 0.0038% | ||||
| Seven Mile Beach NP | 0.015% ± 0.0016% | ||||
| Sharewater | 0.0079% ± 0.0021% | ||||
| K15924 | Glucuronoarabinoxylan endo-1,4-beta-xylanase | Broulee | 0.0065% ± 0.0016% | 0.04 * | |
| Eurobodalla NP | 0.0032% ± 0.0021% | ||||
| Meroo NP | 0.0013% ± 0.00040% | ||||
| Monga NP | 0.0025% ± 0.00049% | ||||
| Murramarang NP | 0.0045% ± 0.0015% | ||||
| Seven Mile Beach NP | 0.0066% ± 0.00074% | ||||
| Sharewater | 0.0037% ± 0.0014% | ||||
| K01811 | Alpha-D-xyloside xylohydrolase | Broulee | 0.025% ± 0.0027% | 0.03 * | |
| Eurobodalla NP | 0.017% ± 0.0027% | ||||
| Meroo NP | 0.015% ± 0.0019% | ||||
| Monga NP | 0.021% ± 0.0021% | ||||
| Murramarang NP | 0.022% ± 0.0017% | ||||
| Seven Mile Beach NP | 0.025% ± 0.00057% | ||||
| Sharewater | 0.012% ± 0.000057% | ||||
| K01805 | Xylose isomerase | Broulee | 0.017% ± 0.0026% | 0.06 | |
| Eurobodalla NP | 0.0053% ± 0.0024% | ||||
| Meroo NP | 0.016% ± 0.0027% | ||||
| Monga NP | 0.012% ± 0.0021% | ||||
| Murramarang NP | 0.014% ± 0.0040% | ||||
| Seven Mile Beach NP | 0.016% ± 0.0013% | ||||
| Sharewater | 0.0063% ± 0.00012% | ||||
| Fibre Degraded | KEGG Orthologue | Enzyme | Mean Relative Abundance (% ± SEM) | S | p Value | |
|---|---|---|---|---|---|---|
| Burnt Habitat | Unburnt Habitat | |||||
| Cellulose | K01179 | Endoglucanase | 0.063% ± 0.0039% | 0.083% ± 0.0062% | 190 | 0.01 * |
| K05350 | Beta-glucosidase | 0.032% ± 0.0016% | 0.033% ± 0.0019% | 146 | 0.89 | |
| Pectin | K18650 | Exo-poly-alphagalacturonosidase | 0.00055% ± 0.000062% | 0.00072% ± 0.000097% | 172 | 0.12 |
| K06113 | Arabinan endo-1,5-alpha-Larabinosidase | 0.024% ± 0.0014% | 0.024% ± 0.0020% | 141 | 0.93 | |
| K01051 | Pectinesterase | 0.0032% ± 0.00051% | 0.0027% ± 0.00044% | 131 | 0.53 | |
| K01728 | Pectate lyase | 0.00017% ± 0.000049% | 0.00013% ± 0.000044% | 131 | 0.53 | |
| Xylan | K01181 | Endo-1,4-beta-xylanase | 0.0095% ± 0.0011% | 0.016% ± 0.0015% | 195 | <0.01 * |
| K15924 | Glucuronoarabinoxylan endo-1,4-beta-xylanase | 0.0028% ± 0.00048% | 0.0057% ± 0.00089% | 191 | <0.01 * | |
| K01811 | Alpha-D-xyloside xylohydrolase | 0.018% ± 0.0014% | 0.023% ± 0.0016% | 178 | 0.59 | |
| K01805 | Xylose isomerase | 0.012% ± 0.0014% | 0.013% ± 0.0064% | 157 | 0.46 | |
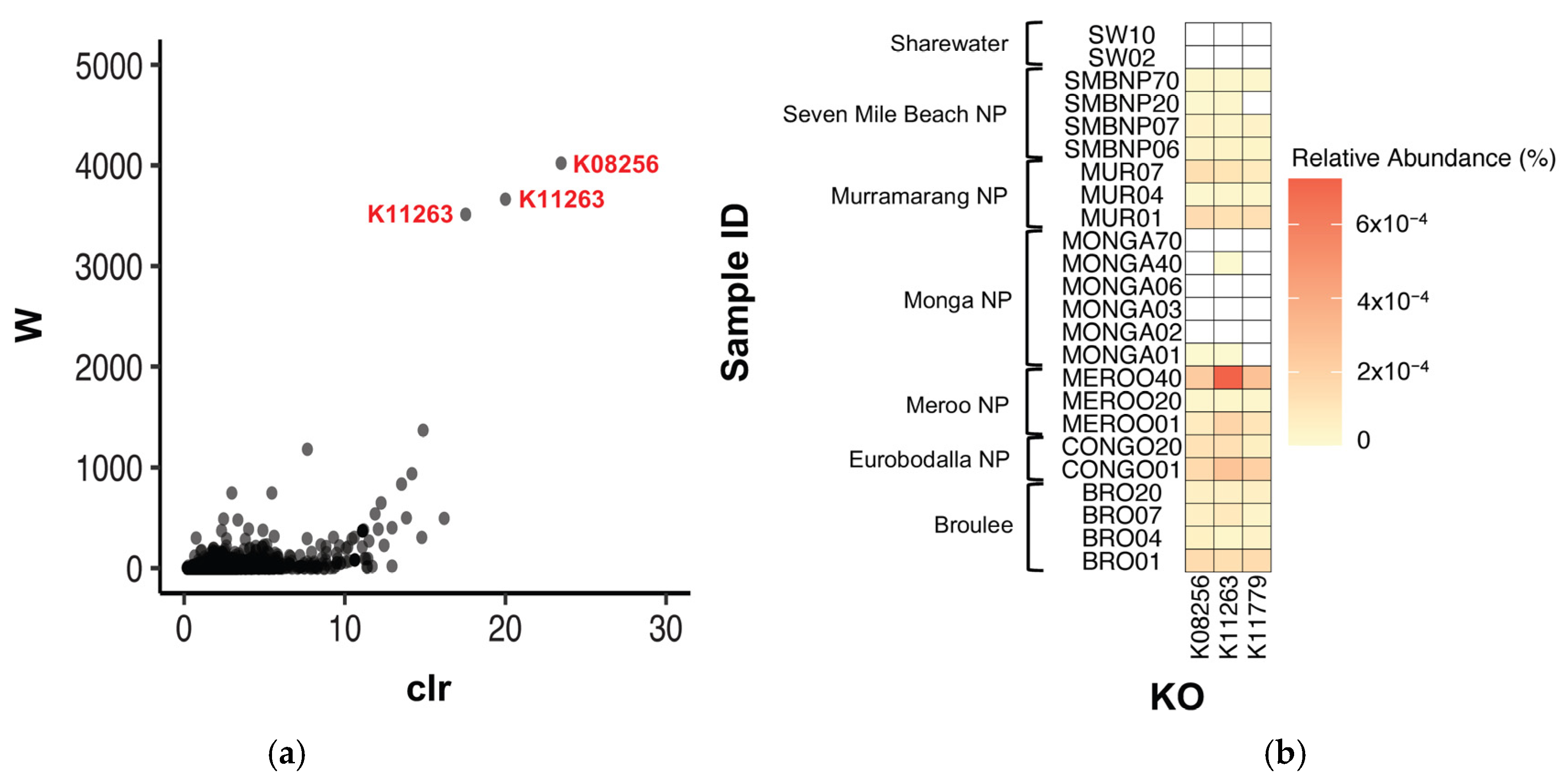
| KO | Name | Pathways | W | Mean Relative Abundance (% ± SEM) | |
|---|---|---|---|---|---|
| K08256 | Phosphatidyl-myo-inositol alpha-mannosyltransferase | Lipoarabinomannan biosynthesis Metabolic pathways | 4388 | Meroo NP | 0.00015 ± 0.00007 |
| Eurobodalla NP | 0.00015 ± 0.000001 | ||||
| Murramarang NP | 0.00011 ± 0.00044 | ||||
| Broulee | 0.00010 ± 0.00003 | ||||
| Seven Mile Beach NP | 0.00003 ± 0.00001 | ||||
| Monga NP | <0.00001% | ||||
| K11779 | F0 synthase | Methane metabolism Metabolic pathways Microbial metabolism in diverse environments | 3894 | Meroo NP | 0.00016 ± 0.00008 |
| Eurobodalla NP | 0.00015 ± 0.000002 | ||||
| Murramarang NP | 0.00011 ± 0.00004 | ||||
| Broulee | 0.00010 ± 0.00003 | ||||
| Seven Mile Beach NP | 0.00003 ± 0.00001 | ||||
| K11263 | Acetyl-CoA/propionyl-CoA carboxylase, biotin carboxylase, biotin carboxyl carrier protein | Fatty acid biosynthesis Valine, leucine and isoleucine degradation Pyruvate metabolism Glyoxylate and dicarboxylate metabolism Propanoate metabolism Metabolic pathways Biosynthesis of secondary metabolites Microbial metabolism in diverse environments Carbon metabolism Fatty acid metabolism | 3863 | Meroo NP | 0.00031 ± 0.00021 |
| Eurobodalla NP | 0.00017 ± 0.00002 | ||||
| Murramarang NP | 0.00011 ± 0.00005 | ||||
| Broulee | 0.00011 ± 0.00004 | ||||
| Seven Mile Beach NP | 0.00003 ± 0.00001 | ||||
| Monga NP | <0.00001 | ||||
References
- West, A.G.; Waite, D.W.; Deines, P.; Bourne, D.G.; Digby, A.; McKenzie, V.J.; Taylor, M.W. The microbiome in threatened species conservation. Biol. Conserv. 2019, 229, 85–98. [Google Scholar] [CrossRef]
- Molina-Torres, G.; Rodriguez-Arrastia, M.; Roman, P.; Sanchez-Labraca, N.; Cardona, D. Stress and the gut microbiota-brain axis. Behav. Pharmacol. 2019, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu. Rev. Med. 2011, 62, 361–380. [Google Scholar] [CrossRef]
- Rogers, G.B.; Keating, D.J.; Young, R.L.; Wong, M.L.; Licinio, J.; Wesselingh, S. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Quinn, R.A.; Debelius, J.; Xu, Z.Z.; Morton, J.; Garg, N.; Jansson, J.K.; Dorrestein, P.C.; Knight, R. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 2016, 535, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.J.; David, A.S.; Menges, E.S.; Searcy, C.A.; Afkhami, M.E. Environmental stress destabilizes microbial networks. ISME J. 2021, 15, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Rocca, J.D.; Simonin, M.; Blaszczak, J.R.; Ernakovich, J.G.; Gibbons, S.M.; Midani, F.S.; Washburne, A.D. The microbiome stress project: Toward a global meta-analysis of environmental stressors and their effects on microbial communities. Front. Microbiol. 2019, 9, 3272. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Trevelline, B.K.; Fontaine, S.S.; Hartup, B.K.; Kohl, K.D. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. Biol. Sci. 2019, 286, 20182448. [Google Scholar] [CrossRef]
- Comport, S.S.; Ward, S.J.; Foley, W.J. Home ranges, time budgets and food-tree use in a high-density tropical population of greater gliders, Petauroides volans minor (Pseudocheiridae: Marsupialia). Wildl. Res. 1996, 23, 401–419. [Google Scholar] [CrossRef]
- Wagner, B.; Baker, P.J.; Nitschke, C.R. The influence of spatial patterns in foraging habitat on the abundance and home range size of a vulnerable arboreal marsupial in southeast Australia. Conserv. Sci. Pract. 2021, 3, e566. [Google Scholar] [CrossRef]
- NSW Office of Environment and Heritage. Southern Greater Glider—Profile. 2023. Available online: https://www.environment.nsw.gov.au/threatenedspeciesapp/profile.aspx?id=20306 (accessed on 26 June 2023).
- NSW Department of Climate Change, Energy, the Environment and Water. Conservation Advice for Petauroides volans (Greater Glider (Southern and Central)); NSW Department of Climate Change, Energy, the Environment and Water: Canberra, ACT, Australia, 2022.
- Barker, C.J.; Gillett, A.; Polkinghorne, A.; Timms, P. Investigation of the koala (Phascolarctos cinereus) hindgut microbiome via 16S pyrosequencing. Vet. Microbiol. 2013, 167, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Alfano, N.; Courtiol, A.; Vielgrader, H.; Timms, P.; Roca, A.L.; Greenwood, A.D. Variation in koala microbiomes within and between individuals: Effect of body region and captivity status. Sci. Rep. 2015, 5, 10189. [Google Scholar] [CrossRef]
- Shiffman, M.E.; Soo, R.M.; Dennis, P.G.; Morrison, M.; Tyson, G.W.; Hugenholtz, P. Gene and genome-centric analyses of koala and wombat fecal microbiomes point to metabolic specialization for Eucalyptus digestion. PeerJ 2017, 5, e4075. [Google Scholar]
- Brice, K.L.; Trivedi, P.; Jeffries, T.C.; Blyton, M.D.J.; Mitchell, C.; Singh, B.K.; Moore, B.D. The Koala (Phascolarctos cinereus) faecal microbiome differs with diet in a wild population. PeerJ 2019, 7, e6534. [Google Scholar] [CrossRef]
- Blyton, M.D.J.; Soo, R.M.; Whisson, D.; Marsh, K.J.; Pascoe, J.; Le Pla, M.; Foley, W.; Hugenholtz, P.; Moore, B.D. Faecal inoculations alter the gastrointestinal microbiome and allow dietary expansion in a wild specialist herbivore, the koala. Anim. Microbiome 2019, 1, 6. [Google Scholar] [CrossRef]
- Blyton, M.D.; Pascoe, J.; Hynes, E.; Soo, R.M.; Hugenholtz, P.; Moore, B.D. The koala gut microbiome is largely unaffected by host translocation but rather influences host diet. Front. Microbiol. 2023, 14, 1085090. [Google Scholar] [CrossRef]
- Bharti, R.; Grimm, D.G. Current challenges and best-practice protocols for microbiome analysis. Brief. Bioinform. 2021, 22, 178–193. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Fox, S.; Pemberton, D.; Hogg, C.; Papenfuss, A.T.; Belov, K. The Tasmanian devil microbiome-implications for conservation and management. Microbiome 2015, 3, 76. [Google Scholar] [CrossRef] [PubMed]
- Burke, C.; Burnard, D.; Polkinghorne, A.; Webb, J.; Huston, W.M. Cloacal and ocular microbiota of the endangered Australian northern quoll. Microorganisms 2018, 6, 68. [Google Scholar] [CrossRef]
- Chong, R.; Grueber, C.E.; Fox, S.; Wise, P.; Barrs, V.R.; Hogg, C.J.; Belov, K. Looking like the locals-gut microbiome changes post-release in an endangered species. Anim. Microbiome 2019, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, R.; Helgen, K.M.; Taggart, D. Signatures of landscape and captivity in the gut microbiota of Southern Hairy-nosed Wombats (Lasiorhinus latifrons). Anim. Microbiome 2021, 3, 4. [Google Scholar] [CrossRef]
- Knipler, M.L.; Gracanin, A.; Mikac, K.M. Conservation genomics of an endangered arboreal mammal following the 2019–2020 Australian megafire. Sci. Rep. 2023, 13, 480. [Google Scholar] [CrossRef]
- State Government of NSW and Department of Planning and Environment. Fire Extent and Severity Mapping (FESM). Accessed from The Sharing and Enabling Environmental Data Portal. 2020. Available online: https://datasets.seed.nsw.gov.au/dataset/33c2ee86-d2f7-4aaf-8c40-76b6d393a35c (accessed on 12 June 2023).
- Gracanin, A.; Pearce, A.; Hofman, M.; Knipler, M.; Mikac, K.M. Greater glider (Petauroides volans) live capture methods. Aust. Mammal. 2021, 44, 280–286. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 326–349. [Google Scholar] [CrossRef]
- Jaccard, P. Nouvelles recherches sur la distribution florale. Bull. Soc. Vaud. Sci. Nat. 1908, 44, 223–270. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Raplee, I.; Walker, L.; Xu, L.; Surathu, A.; Chockalingam, A.; Stewart, S.; Han, X.; Rouse, R.; Li, Z. Emergence of nosocomial associated opportunistic pathogens in the gut microbiome after antibiotic treatment. Antimicrob. Resist. Infect. Control 2021, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, R.P.; Lambert, M.J. Food selection by the greater glider, Petauroides volans: Is foliar nitrogen a determinant of habitat quality? Aust. Wildl. Res. 1990, 17, 285–299. [Google Scholar] [CrossRef]
- Zhang, L.; Zhan, H.; Xu, W.; Yan, S.; Ng, S.C. The role of gut mycobiome in health and diseases. Therap. Adv. Gastroenterol. 2021, 14, 17562848211047130. [Google Scholar] [CrossRef] [PubMed]
- Honneffer, J.B.; Minamoto, Y.; Suchodolski, J.S. Microbiota alterations in acute and chronic gastrointestinal inflammation of cats and dogs. World J. Gastroenterol. 2014, 20, 16489–16497. [Google Scholar] [CrossRef]
- AlShawaqfeh, M.K.; Wajid, B.; Minamoto, Y.; Markel, M.; Lidbury, J.A.; Steiner, J.M.; Serpedin, E.; Suchodolski, J.S. A dysbiosis index to assess microbial changes in fecal samples of dogs with chronic inflammatory enteropathy. FEMS Microbiol. Ecol. 2017, 93, fix136. [Google Scholar] [CrossRef]
- Felix, A.P.; Souza, C.M.M.; de Oliveira, S.G. Biomarkers of gastrointestinal functionality in dogs: A systematic review and meta-analysis. Anim. Feed Sci. Technol. 2022, 283, 115183. [Google Scholar] [CrossRef]
- Delport, T.C.; Power, M.L.; Harcourt, R.G.; Webster, K.N.; Tetu, S.G. Colony Location and Captivity Influence the Gut Microbial Community Composition of the Australian Sea Lion (Neophoca cinerea). Appl. Environ. Microbiol. 2016, 82, 3440–3449. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Q.; Song, J.; Xin, J.; Zhang, S.; Lei, Y.; Yang, Y.; Xie, P.; Suo, H. Comparison of Gut Microbiota of Yaks From Different Geographical Regions. Front. Microbiol. 2021, 12, 666940. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, C.; Li, G.; Yi, X. The influence of species identity and geographic locations on gut microbiota of small rodents. Front. Microbiol. 2022, 13, 983660. [Google Scholar] [CrossRef]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef]
- Moeller, A.H.; Suzuki, T.A.; Lin, D.; Lacey, E.A.; Wasser, S.K.; Nachman, M.W. Dispersal limitation promotes the diversification of the mammalian gut microbiota. Proc. Natl. Acad. Sci. USA 2017, 114, 13768–13773. [Google Scholar] [CrossRef] [PubMed]
- Senghor, B.; Sokhna, C.; Ruimy, R.; Lagier, J.-C. Gut microbiota diversity according to dietary habits and geographical provenance. Hum. Microbiome J. 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Lemieux-Labonte, V.; Vigliotti, C.; Tadic, Z.; Wehrle, B.; Lopez, P.; Bapteste, E.; Lapointe, F.J.; German, D.P.; Herrel, A. Proximate drivers of population-level lizard gut microbial diversity: Impacts of diet, insularity, and local environment. Microorganisms 2022, 10, 1550. [Google Scholar] [CrossRef]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef]
- Makki, K.; Deehan, E.C.; Walter, J.; Backhed, F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef]
- NSW Office of Environment and Heritage. South Coast Sands Dry Sclerophyll Forests. 2023. Available online: https://www.environment.nsw.gov.au/threatenedspeciesapp/VegClass.aspx?vegClassName=South%20Coast%20Sands%20Dry%20Sclerophyll%20Forests (accessed on 22 July 2023).
- Vinson, S.G.; Johnson, A.P.; Mikac, K.M. Current estimates and vegetation preferences of an endangered population of the vulnerable greater glider at Seven Mile Beach National Park. Austral. Ecol. 2021, 46, 303–314. [Google Scholar] [CrossRef]
- Fackelmann, G.; Gillingham, M.A.; Schmid, J.; Heni, A.C.; Wilhelm, K.; Schwensow, N.; Sommer, S. Human encroachment into wildlife gut microbiomes. Commun. Biol. 2021, 4, 800. [Google Scholar] [CrossRef]
- Wasimuddin; Malik, H.; Ratovonamana, Y.R.; Rakotondranary, S.J.; Ganzhorn, J.U.; Sommer, S. Anthropogenic Disturbance Impacts Gut Microbiome Homeostasis in a Malagasy Primate. Front. Microbiol. 2022, 13, 911275. [Google Scholar] [CrossRef]
- Lobato-Bailon, L.; Garcia-Ulloa, M.; Santos, A.; Guixe, D.; Camprodon, J.; Florensa-Rius, X.; Molleda, R.; Manzano, R.; Ribas, M.P.; Espunyes, J.; et al. The fecal bacterial microbiome of the Kuhl’s pipistrelle bat (Pipistrellus kuhlii) reflects landscape anthropogenic pressure. Anim. Microbiome 2023, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Heni, A.C.; Fackelmann, G.; Eibner, G.; Kreinert, S.; Schmid, J.; Schwensow, N.I.; Wiegand, J.; Wilhelm, K.; Sommer, S. Wildlife gut microbiomes of sympatric generalist species respond differently to anthropogenic landscape disturbances. Anim. Microbiome 2023, 5, 22. [Google Scholar] [CrossRef]
- NSW Department of Planning and Environment. Greater Glider (Petauroides volans) Population in the Eurobodalla Local Government Area—Endangered Population Listing. 2007. Available online: https://www.environment.nsw.gov.au/topics/animals-and-plants/threatened-species/nsw-threatened-species-scientific-committee/determinations/final-determinations/2004-2007/greater-glider-petauroides-volans-endangered-population-listing (accessed on 22 July 2023).
- Tung, J.; Barreiro, L.B.; Burns, M.B.; Grenier, J.C.; Lynch, J.; Grieneisen, L.E.; Altmann, J.; Alberts, S.C.; Blekhman, R.; Archie, E.A. Social networks predict gut microbiome composition in wild baboons. Elife 2015, 4, e05224. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.H.; Foerster, S.; Wilson, M.L.; Pusey, A.E.; Hahn, B.H.; Ochman, H. Social behavior shapes the chimpanzee pan-microbiome. Sci. Adv. 2016, 2, e1500997. [Google Scholar] [CrossRef] [PubMed]
- Perofsky, A.C.; Ancel Meyers, L.; Abondano, L.A.; Di Fiore, A.; Lewis, R.J. Social groups constrain the spatiotemporal dynamics of wild sifaka gut microbiomes. Mol. Ecol. 2021, 30, 6759–6775. [Google Scholar] [CrossRef]
- Henry, S. Social organisation of the greater glider (Petauroides volans) in Victoria. In Possums and Gliders; Smith, A., Hume, I., Eds.; Surrey Beatty & Sons: Sydney, NSW, Australia, 1984; pp. 221–228. [Google Scholar]
- Norton, T.W. Ecology of Greater Gliders, Petauroides volans Kerr 1792, in Relation to Variations in Habitat Quality in Eucalypt Forests in South-East New South Wales; The Australian National University: Canberra, ACT, Australia, 1988. [Google Scholar]
- Gulino, L.M.; Ouwerkerk, D.; Kang, A.Y.; Maguire, A.J.; Kienzle, M.; Klieve, A.V. Shedding light on the microbial community of the macropod foregut using 454-amplicon pyrosequencing. PLoS ONE 2013, 8, e61463. [Google Scholar] [CrossRef]
- Cork, S.J.; Hume, I.; Dawson, T. Digestion and metabolism of a natural foliar diet (Eucalyptus punctata) by an arboreal marsupial, the koala (Phascolarctos cinereus). J. Comp. Physiol. 1983, 153, 181–190. [Google Scholar] [CrossRef]
- Chilcott, M.; Hume, I. Digestion of Eucalyptus andrewsii foliage by the common ringtail possum, Pseudocheirus peregrinus. Aust. J. Zool. 1984, 32, 605–613. [Google Scholar] [CrossRef]
- Foley, W. Digestion and energy metabolism in a small arboreal marsupial, the Greater Glider (Petauroides volans), fed high-terpene Eucalyptus foliage. J. Comp. Physiol. B 1987, 157, 355–362. [Google Scholar] [CrossRef]
- Stojanov, S.; Berlec, A.; Strukelj, B. The influence of probiotics on the Firmicutes/Bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.-H.; Zhu, C.-X.; Quan, Y.-S.; Yang, Z.-Y.; Wu, S.; Luo, W.-W.; Tan, B.; Wang, X.-Y. Relationship between intestinal microbiota and ulcerative colitis: Mechanisms and clinical application of probiotics and fecal microbiota transplantation. World J. Gastroenterol. 2018, 24, 5. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Scarpellini, E.; Colica, C.; Boccuto, L.; Salehi, B.; Sharifi-Rad, J.; Aiello, V.; Romano, B.; De Lorenzo, A.; Izzo, A.A.; et al. Gut microbiota and obesity: A role for probiotics. Nutrients 2019, 11, 2690. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Meehan, C.J.; Beiko, R.G. A phylogenomic view of ecological specialization in the Lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol. Evol. 2014, 6, 703–713. [Google Scholar] [CrossRef]
- Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]
- Kabel, M.A.; Yeoman, C.J.; Han, Y.; Dodd, D.; Abbas, C.A.; de Bont, J.A.; Morrison, M.; Cann, I.K.; Mackie, R.I. Biochemical characterization and relative expression levels of multiple carbohydrate esterases of the xylanolytic rumen bacterium Prevotella ruminicola 23 grown on an ester-enriched substrate. Appl. Environ. Microbiol. 2011, 77, 5671–5681. [Google Scholar] [CrossRef]
- Thoetkiattikul, H.; Mhuantong, W.; Laothanachareon, T.; Tangphatsornruang, S.; Pattarajinda, V.; Eurwilaichitr, L.; Champreda, V. Comparative analysis of microbial profiles in cow rumen fed with different dietary fiber by tagged 16S rRNA gene pyrosequencing. Curr. Microbiol. 2013, 67, 130–137. [Google Scholar] [CrossRef]
- Kovatcheva-Datchary, P.; Nilsson, A.; Akrami, R.; Lee, Y.S.; De Vadder, F.; Arora, T.; Hallen, A.; Martens, E.; Björck, I.; Bäckhed, F. Dietary fiber-induced improvement in glucose metabolism is associated with increased abundance of Prevotella. Cell Metab. 2015, 22, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Zoelzer, F.; Burger, A.L.; Dierkes, P.W. Unraveling differences in fecal microbiota stability in mammals: From high variable carnivores and consistently stable herbivores. Anim. Microbiome 2021, 3, 77. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Gu, W.; Lee, I.A.; Joh, E.H.; Kim, D.H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- La Reau, A.J.; Suen, G. The Ruminococci: Key symbionts of the gut ecosystem. J. Microbiol. 2018, 56, 199–208. [Google Scholar] [CrossRef]
- Kartzinel, T.R.; Hsing, J.C.; Musili, P.M.; Brown, B.R.P.; Pringle, R.M. Covariation of diet and gut microbiome in African megafauna. Proc. Natl. Acad. Sci. USA 2019, 116, 23588–23593. [Google Scholar] [CrossRef]
- Asma, Z.; Sylvie, C.; Laurent, C.; Jérôme, M.; Christophe, K.; Olivier, B.; Annabelle, T.-M.; Francis, E. Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets. FEMS Microbiol. Ecol. 2013, 83, 504–514. [Google Scholar]
- Wang, H.; He, Y.; Li, H.; Wu, F.; Qiu, Q.; Niu, W.; Gao, Z.; Su, H.; Cao, B. Rumen fermentation, intramuscular fat fatty acid profiles and related rumen bacterial populations of Holstein bulls fed diets with different energy levels. Appl. Microbiol. Biotechnol. 2019, 103, 4931–4942. [Google Scholar] [CrossRef]
- Crognale, S.; Massimi, A.; Sbicego, M.; Braguglia, C.M.; Gallipoli, A.; Gazzola, G.; Gianico, A.; Tonanzi, B.; Di Pippo, F.; Rossetti, S. Ecology of food waste chain-elongating microbiome. Front. Bioeng. Biotechnol. 2023, 11, 1157243. [Google Scholar] [CrossRef]
- Fan, P.; Kim, M.; Liu, G.; Zhai, Y.; Liu, T.; Driver, J.D.; Jeong, K.C. The Gut Microbiota of Newborn Calves and Influence of Potential Probiotics on Reducing Diarrheic Disease by Inhibition of Pathogen Colonization. Front. Microbiol. 2021, 12, 772863. [Google Scholar] [CrossRef]
- Zeng, B.; Zhang, S.; Xu, H.; Kong, F.; Yu, X.; Wang, P.; Yang, M.; Li, D.; Zhang, M.; Ni, Q. Gut microbiota of Tibetans and Tibetan pigs varies between high and low altitude environments. Microbiol. Res. 2020, 235, 126447. [Google Scholar] [CrossRef]
- Li, H.; Zhou, R.; Zhu, J.; Huang, X.; Qu, J. Environmental filtering increases with elevation for the assembly of gut microbiota in wild pikas. Microb. Biotechnol. 2019, 12, 976–992. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yao, Y.; Li, D.; Xu, H.; Wu, J.; Wen, A.; Xie, M.; Ni, Q.; Zhang, M.; Peng, G.; et al. Characterization of the Gut Microbiota in Six Geographical Populations of Chinese Rhesus Macaques (Macaca mulatta), Implying an Adaptation to High-Altitude Environment. Microb. Ecol. 2018, 76, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Ward, D.V.; Pasolli, E.; Tolio, T.; Zolfo, M.; Asnicar, F.; Truong, D.T.; Tett, A.; Morrow, A.L.; Segata, N. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods 2016, 13, 435–438. [Google Scholar] [CrossRef]
- Ebringerová, A.; Heinze, T. Xylan and xylan derivatives—Biopolymers with valuable properties, 1. Naturally occurring xylans structures, isolation procedures and properties. Macromol. Rapid Commun. 2000, 21, 542–556. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, B.; Lu, T.; Huang, Z. Metagenomic Analysis of the Fecal Microbiomes of Wild Asian Elephants Reveals Microflora and Enzymes that Mainly Digest Hemicellulose. J. Microbiol. Biotechnol. 2019, 29, 1255–1265. [Google Scholar] [CrossRef]
- Horn, S.J.; Vaaje-Kolstad, G.; Westereng, B.; Eijsink, V.G. Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 2012, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Boldrin, F.; Ventura, M.; Degiacomi, G.; Ravishankar, S.; Sala, C.; Svetlikova, Z.; Ambady, A.; Dhar, N.; Kordulakova, J.; Zhang, M.; et al. The phosphatidyl-myo-inositol mannosyltransferase PimA is essential for Mycobacterium tuberculosis growth in vitro and in vivo. J. Bacteriol. 2014, 196, 3441–3451. [Google Scholar] [CrossRef]
- Fitzgerald, S.D.; Kaneene, J.B. Wildlife reservoirs of bovine tuberculosis worldwide: Hosts, pathology, surveillance, and control. Vet. Pathol. 2013, 50, 488–499. [Google Scholar] [CrossRef]
- Tschopp, R.; Schelling, E.; Hattendorf, J.; Aseffa, A.; Zinsstag, J. Risk factors of bovine tuberculosis in cattle in rural livestock production systems of Ethiopia. Prev. Vet. Med. 2009, 89, 205–211. [Google Scholar] [CrossRef]
- Malone, K.M.; Gordon, S.V. Mycobacterium tuberculosis Complex Members Adapted to Wild and Domestic Animals. Adv. Exp. Med. Biol. 2017, 1019, 135–154. [Google Scholar] [CrossRef]
- Olea-Popelka, F.; Muwonge, A.; Perera, A.; Dean, A.S.; Mumford, E.; Erlacher-Vindel, E.; Forcella, S.; Silk, B.J.; Ditiu, L.; El Idrissi, A.; et al. Zoonotic tuberculosis in human beings caused by Mycobacterium bovis—A call for action. Lancet Infect. Dis. 2017, 17, e21–e25. [Google Scholar] [CrossRef] [PubMed]
- Nugent, G. Maintenance, spillover and spillback transmission of bovine tuberculosis in multi-host wildlife complexes: A New Zealand case study. Vet. Microbiol. 2011, 151, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Nugent, G.; Buddle, B.M.; Knowles, G. Epidemiology and control of Mycobacterium bovis infection in brushtail possums (Trichosurus vulpecula), the primary wildlife host of bovine tuberculosis in New Zealand. N. Z. Vet. J. 2015, 63 (Suppl. 1), 28–41. [Google Scholar] [CrossRef]
- More, S.J.; Radunz, B.; Glanville, R.J. Lessons learned during the successful eradication of bovine tuberculosis from Australia. Vet. Rec. 2015, 177, 224–232. [Google Scholar] [CrossRef]
- Harrison, S.; Hastings, A. Genetic and evolutionary consequences of metapopulation structure. Trends Ecol. Evol. 1996, 11, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Dudash, M.R.; Fenster, C.B. Inbreeding and outbreeding depression in fragmeneted populations. In Genetics, Demography and Viability of Fragmented Populations; Young, A.G., Clarke, G.M., Eds.; Cambridge University Press: Cambridge, UK, 2000; pp. 35–54. [Google Scholar]
- Keller, L. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002, 17, 230–241. [Google Scholar] [CrossRef]
- Sommer, S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2005, 2, 16. [Google Scholar] [CrossRef]
- Eurobodalla Shire Council Heritage Advisory Committee. Eurobodalla Dairy Industry Heritage Scoping Study; NSW Heritage Council: Parramatta, NSW, Australia, 2006.
- Lindsay, S.A.; Gray, R. A novel presentation of tuberculosis with intestinal perforation in a free-ranging Australian sea lion (Neophoca cinerea). J. Wildl. Dis. 2021, 57, 220–224. [Google Scholar] [CrossRef]
- Spor, A.; Koren, O.; Ley, R. Unravelling the effects of the environment and host genotype on the gut microbiome. Nat. Rev. Microbiol. 2011, 9, 279–290. [Google Scholar] [CrossRef]
- Ward, M.; Tulloch, A.I.T.; Radford, J.Q.; Williams, B.A.; Reside, A.E.; Macdonald, S.L.; Mayfield, H.J.; Maron, M.; Possingham, H.P.; Vine, S.J.; et al. Impact of 2019–2020 mega-fires on Australian fauna habitat. Nat. Ecol. Evol. 2020, 4, 1321–1326. [Google Scholar] [CrossRef]
- Perry, T.; Lu, A.; McKelvey, M.W.; Rismiller, P.D.; Grützner, F. Bushfire alters the gut microbiome in endangered Kangaroo Island echidnas (Tachyglossus aculeatus multiaculeatus). bioRxiv 2023. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Gaskins, H.R.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Barelli, C.; Albanese, D.; Donati, C.; Pindo, M.; Dallago, C.; Rovero, F.; Cavalieri, D.; Tuohy, K.M.; Hauffe, H.C.; De Filippo, C. Habitat fragmentation is associated to gut microbiota diversity of an endangered primate: Implications for conservation. Sci. Rep. 2015, 5, 14862. [Google Scholar] [CrossRef] [PubMed]
- Jumas-Bilak, E.; Roudiere, L.; Marchandin, H. Description of ‘Synergistetes’ phyl. nov. and emended description of the phylum ‘Deferribacteres’ and of the family Syntrophomonadaceae, phylum ‘Firmicutes’. Int. J. Syst. Evol. Microbiol. 2009, 59, 1028–1035. [Google Scholar] [CrossRef]
- Bhandari, V.; Gupta, R.S. Molecular signatures for the phylum Synergistetes and some of its subclades. Antonie Van Leeuwenhoek 2012, 102, 517–540. [Google Scholar] [CrossRef]
- Gibson, K.M.; Nguyen, B.N.; Neumann, L.M.; Miller, M.; Buss, P.; Daniels, S.; Ahn, M.J.; Crandall, K.A.; Pukazhenthi, B. Gut microbiome differences between wild and captive black rhinoceros—Implications for rhino health. Sci. Rep. 2019, 9, 7570. [Google Scholar] [CrossRef]
- Yao, R.; Xu, L.; Hu, T.; Chen, H.; Qi, D.; Gu, X.; Yang, X.; Yang, Z.; Zhu, L. The “wildness” of the giant panda gut microbiome and its relevance to effective translocation. Glob. Ecol. Conserv. 2019, 18, e00644. [Google Scholar] [CrossRef]
- Wasimuddin; Menke, S.; Melzheimer, J.; Thalwitzer, S.; Heinrich, S.; Wachter, B.; Sommer, S. Gut microbiomes of free-ranging and captive Namibian cheetahs: Diversity, putative functions and occurrence of potential pathogens. Mol. Ecol. 2017, 26, 5515–5527. [Google Scholar] [CrossRef]
- Bradstock, R.A.; Bedward, M.; Gill, A.M.; Cohn, J.S. Which mosaic? A landscape ecological approach for evaluating interactions between fire regimes, habitat and animals. Wildl. Res. 2005, 32, 409–423. [Google Scholar] [CrossRef]
- May-Stubbles, J.C.; Gracanin, A.; Mikac, K.M.; Robinson, N. Increasing fire severity negatively affects greater glider density. Wildl. Res. 2022, 49, 709–718. [Google Scholar] [CrossRef]
- Berry, L.E.; Driscoll, D.A.; Stein, J.A.; Blanchard, W.; Banks, S.C.; Bradstock, R.A.; Lindenmayer, D.B. Identifying the location of fire refuges in wet forest ecosystems. Ecol. Appl. 2015, 25, 2337–2348. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.P.; Neville, B.A.; Forster, S.C.; Lawley, T.D. Transmission of the gut microbiota: Spreading of health. Nat. Rev. Microbiol. 2017, 15, 531–543. [Google Scholar] [CrossRef] [PubMed]
- State Government of NSW and Department of Planning and Environment. NSW State Vegetation Type Map. Accessed from The Sharing and Enabling Environmental Data Portal. 2022. Available online: https://datasets.seed.nsw.gov.au/dataset/95437fbd-2ef7-44df-8579-d7a64402d42d (accessed on 13 September 2023).






| Number of Observed Features (ASVs) | Chao1 | Shannon | |
|---|---|---|---|
| Location | 15.52 * | 15.52 * | 16.18 * |
| Burn status of site | 2.69 | 2.69 | 1.37 |
| Sex | 1.66 | 1.66 | 0.31 |
| Month collected | 8.35 | 8.35 | 5.28 |
| Bray–Curtis Distance | Jaccard Distance | Unweighted UniFrac Distance | Weighted UniFrac Distance | |
|---|---|---|---|---|
| Location | 3.64 * | 3.33 * | 2.14 * | 2.86 * |
| Burn status of site | 2.60 * | 2.85 * | 2.33 * | 0.81 |
| Sex | 0.84 | 0.89 | 0.87 | 0.76 |
| Taxonomic Level | Taxa | Mean Relative Abundance (% ± SEM) | p Value | |
|---|---|---|---|---|
| Burnt Habitat | Unburnt Habitat | |||
| Phylum | Firmicutes | 61.01 ± 2.23 | 66.08 ± 8.96 | 0.23 |
| Bacteroidota | 24.15 ± 2.21 | 22.03 ± 9.74 | 0.70 | |
| Proteobacteria | 4.45 ± 2.59 | 2.46 ± 1.47 | 0.33 | |
| Verrucomicrobiota | 2.75 ± 0.63 | 4.09 ± 2.38 | 0.15 | |
| Synergistota | 3.67 ± 0.52 | 2.01 ± 0.57 | 0.03 * | |
| Family | Lachnospiraceae | 32.78 ± 1.69 | 33.40 ± 3.35 | 0.70 |
| Unclassified Firmicutes | 13.10 ± 1.31 | 11.15 ± 1.91 | 0.23 | |
| Erysipelatoclostridiaceae | 8.54 ± 2.36 | 10.87 ± 3.10 | 0.36 | |
| Prevotellaceae | 10.13 ± 1.24 | 10.45 ± 1.51 | 0.84 | |
| Rikenellaceae | 10.85 ± 2.18 | 8.38 ± 2.04 | 0.62 | |
| Genus | Unclassified Lachnospiraceae | 30.98 ± 1.71 | 31.16 ± 3.41 | 0.77 |
| Unclassified Firmicutes | 13.10 ± 1.31 | 11.15 ± 1.91 | 0.23 | |
| Rickenellaceae RC9 group | 10.77 ± 2.19 | 8.28 ± 2.04 | 0.62 | |
| Erysipelatoclostridiaceae UCG04 | 8.54 ± 2.36 | 10.86 ± 9.81 | 0.36 | |
| Unclassified Prevotellaceae | 5.15 ± 0.57 | 6.57 ± 1.18 | 0.21 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clough, J.; Schwab, S.; Mikac, K. Gut Microbiome Profiling of the Endangered Southern Greater Glider (Petauroides volans) after the 2019–2020 Australian Megafire. Animals 2023, 13, 3583. https://doi.org/10.3390/ani13223583
Clough J, Schwab S, Mikac K. Gut Microbiome Profiling of the Endangered Southern Greater Glider (Petauroides volans) after the 2019–2020 Australian Megafire. Animals. 2023; 13(22):3583. https://doi.org/10.3390/ani13223583
Chicago/Turabian StyleClough, Jordyn, Sibylle Schwab, and Katarina Mikac. 2023. "Gut Microbiome Profiling of the Endangered Southern Greater Glider (Petauroides volans) after the 2019–2020 Australian Megafire" Animals 13, no. 22: 3583. https://doi.org/10.3390/ani13223583
APA StyleClough, J., Schwab, S., & Mikac, K. (2023). Gut Microbiome Profiling of the Endangered Southern Greater Glider (Petauroides volans) after the 2019–2020 Australian Megafire. Animals, 13(22), 3583. https://doi.org/10.3390/ani13223583