
Utilizing Variants Identified with Multiple Genome-Wide Association Study Methods Optimizes Genomic Selection for Growth Traits in Pigs
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Phenotype Data
2.2. Genotype Data
2.3. Imputation of SNP Chips
2.4. Statistical Models for GWAS
2.4.1. Mixed Linear Model
2.4.2. Meta-Analysis
2.4.3. Bayesian Model
2.5. Candidate Genes Annotation
2.6. Statistical Models for GS
2.6.1. GBLUP
2.6.2. Two-Kernel Based GBLUP
2.7. Evaluation of the Accuracy of GS
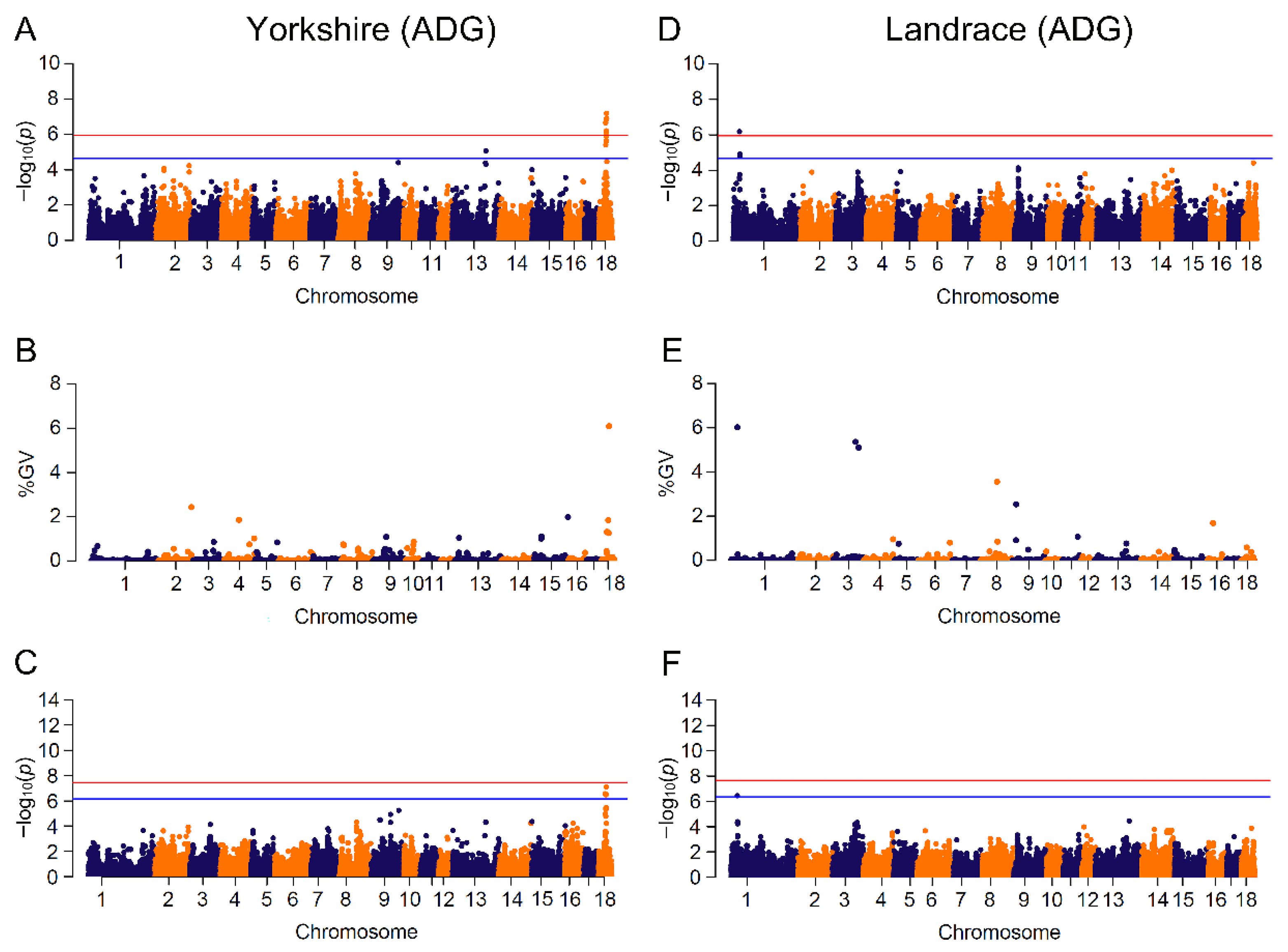
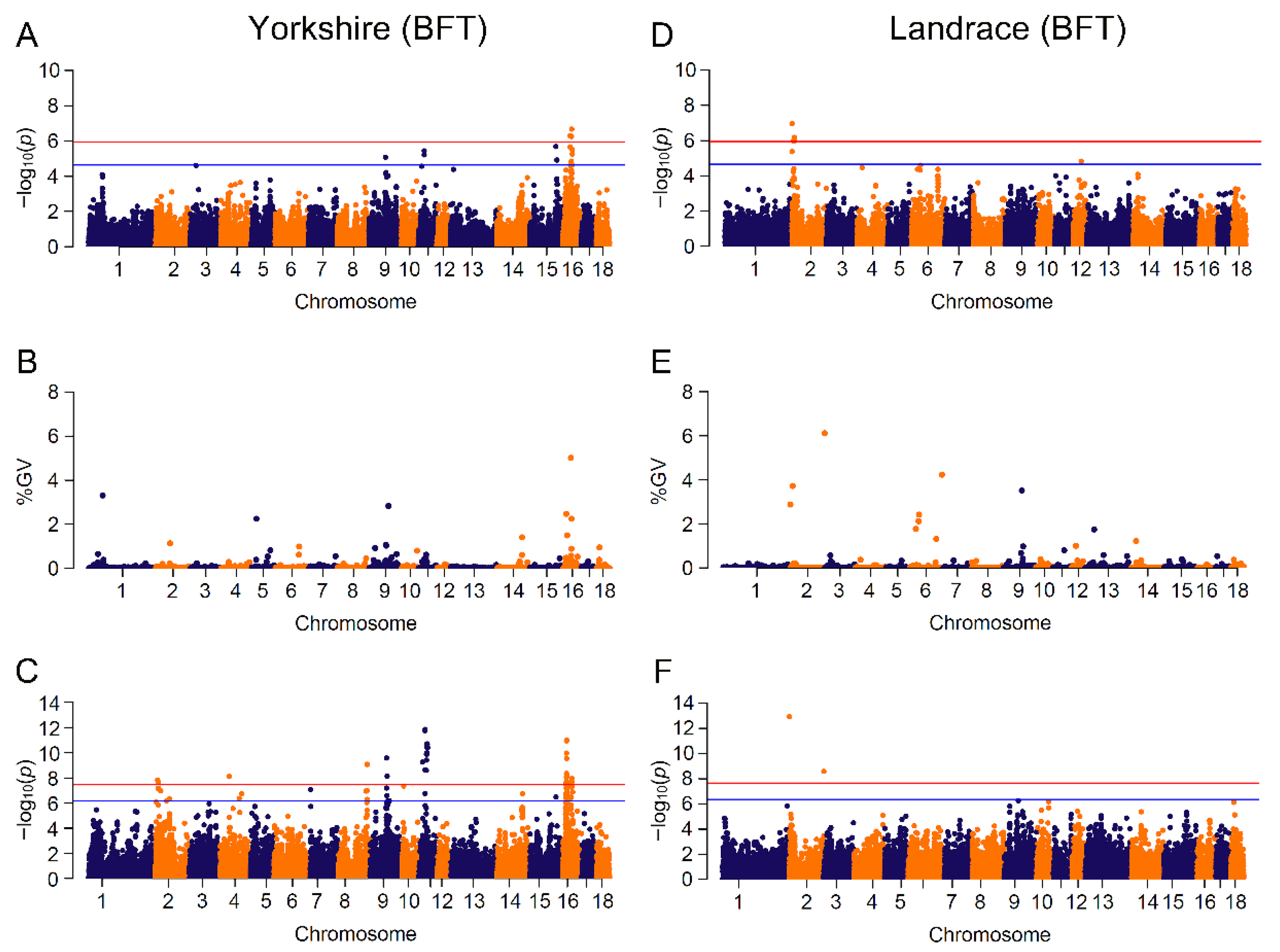
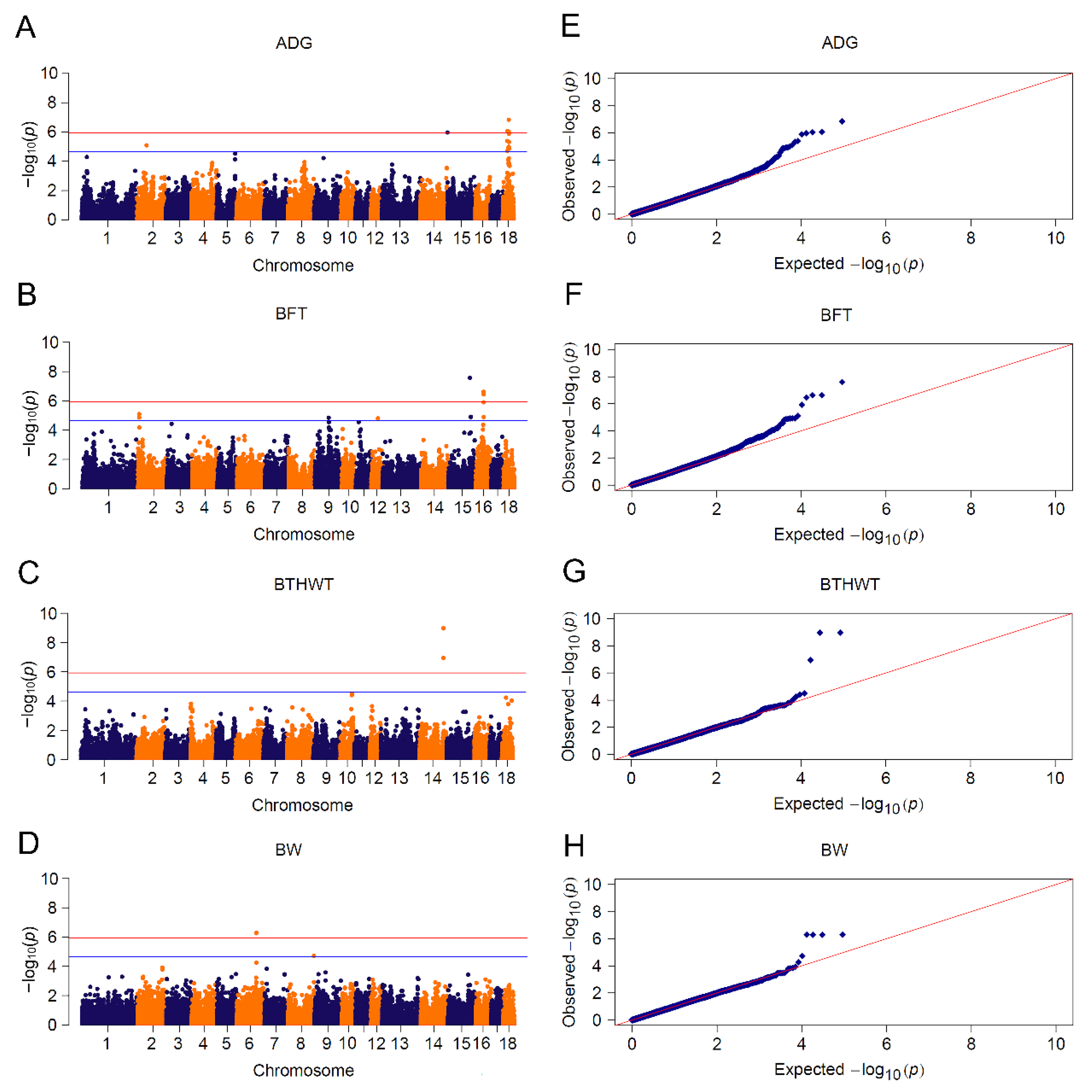
3. Results and Discussion
3.1. Phenotypic Statistics and Heritability Estimation
3.2. Population Structure Analysis
3.3. GWAS Based on Mixed Linear Model and Bayesian Model
3.4. Meta-Analyses
3.5. Genomic Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Kemp, R.A.; Stothard, P.; Wang, Z.; Boddicker, N.; Krivushin, K.; Dekkers, J.; Plastow, G. Genomic evaluation of feed efficiency component traits in Duroc pigs using 80K, 650K and whole-genome sequence variants. Genet. Sel. Evol. 2018, 50, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, T.; Goddard, M. Accurate Prediction of Genetic Values for Complex Traits by Whole-Genome Resequencing. Genetics 2010, 185, 623–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druet, T.; Macleod, I.M.; Hayes, B.J. Toward genomic prediction from whole-genome sequence data: Impact of sequencing design on genotype imputation and accuracy of predictions. Heredity 2014, 112, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iheshiulor, O.O.M.; Woolliams, J.A.; Yu, X.; Wellmann, R.; Meuwissen, T.H.E. Within- and across-breed genomic prediction using whole-genome sequence and single nucleotide polymorphism panels. Genet. Sel. Evol. 2016, 48, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.-Y.; Toghiani, S.; Ling, A.; Aggrey, S.E.; Rekaya, R. High density marker panels, SNPs prioritizing and accuracy of genomic selection. BMC Genet. 2018, 19, 4. [Google Scholar] [CrossRef] [Green Version]
- Ni, G.; Cavero, D.; Fangmann, A.; Erbe, M.; Simianer, H. Whole-genome sequence-based genomic prediction in laying chickens with different genomic relationship matrices to account for genetic architecture. Genet. Sel. Evol. 2017, 49, 8. [Google Scholar] [CrossRef] [Green Version]
- Teissier, M.; Sanchez, M.P.; Boussaha, M.; Barbat, A.; Hoze, C.; Robert-Granie, C.; Croiseau, P. Use of meta-analyses and joint analyses to select variants in whole genome sequences for genomic evaluation: An application in milk production of French dairy cattle breeds. J. Dairy Sci. 2018, 101, 3126–3139. [Google Scholar] [CrossRef] [Green Version]
- Raymond, B.; Bouwman, A.C.; Wientjes, Y.C.J.; Schrooten, C.; Houwing-Duistermaat, J.; Veerkamp, R.F. Genomic prediction for numerically small breeds, using models with pre-selected and differentially weighted markers. Genet. Sel. Evol. 2018, 50, 49. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, I.M.; Bowman, P.J.; Vander Jagt, C.J.; Haile-Mariam, M.; Kemper, K.E.; Chamberlain, A.J.; Schrooten, C.; Hayes, B.J.; Goddard, M.E. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genom. 2016, 17, 144. [Google Scholar] [CrossRef] [Green Version]
- Corredor, F.-A.; Sanglard, L.P.; Leach, R.J.; Ross, J.W.; Keating, A.F.; Serão, N.V.L. Genetic and genomic characterization of vulva size traits in Yorkshire and Landrace gilts. BMC Genet. 2020, 21, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, D.N.; Janss, L.L.G.; Jensen, J.; Kadarmideen, H.N. SNP annotation-based whole genomic prediction and selection: An application to feed efficiency and its component traits in pigs. J. Anim. Sci. 2015, 93, 2056–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brøndum, R.F.; Su, G.; Janss, L.; Sahana, G.; Guldbrandtsen, B.; Boichard, D.; Lund, M.S. Quantitative trait loci markers derived from whole genome sequence data increases the reliability of genomic prediction. J. Dairy Sci. 2015, 98, 4107–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozalo-Marcilla, M.; Buntjer, J.; Johnsson, M.; Batista, L.; Diez, F.; Werner, C.R.; Chen, C.-Y.; Gorjanc, G.; Mellanby, R.J.; Hickey, J.M.; et al. Genetic architecture and major genes for backfat thickness in pig lines of diverse genetic backgrounds. Genet. Sel. Evol. 2021, 53, 76. [Google Scholar] [CrossRef]
- Qiao, R.; Gao, J.; Zhang, Z.; Li, L.; Xie, X.; Fan, Y.; Cui, L.; Ma, J.; Ai, H.; Ren, J.; et al. Genome-wide association analyses reveal significant loci and strong candidate genes for growth and fatness traits in two pig populations. Genet. Sel. Evol. 2015, 47, 17. [Google Scholar] [CrossRef] [Green Version]
- Sahana, G.; Guldbrandtsen, B.; Janss, L.; Lund, M.S. Comparison of association mapping methods in a complex pedigreed population. Genet. Epidemiol. 2010, 34, 455–462. [Google Scholar] [CrossRef]
- Dashab, G.R.; Kadri, N.K.; Shariati, M.M.; Sahana, G. Comparison of linear mixed model analysis and genealogy-based haplotype clustering with a Bayesian approach for association mapping in a pedigreed population. BMC Proc. 2012, 6, S4. [Google Scholar] [CrossRef] [Green Version]
- Akey, J.; Jin, L.; Xiong, M. Haplotypes vs single marker linkage disequilibrium tests: What do we gain? Eur. J. Hum. Genet. 2001, 9, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Morris, N.J.; Schaid, D.J.; Elston, R.C. Power of Single- vs. Multi-Marker Tests of Association. Genet. Epidemiol. 2012, 36, 480–487. [Google Scholar] [CrossRef] [Green Version]
- Kemper, K.E.; Reich, C.M.; Bowman, P.J.; vander Jagt, C.J.; Chamberlain, A.J.; Mason, B.A.; Hayes, B.J.; Goddard, M.E. Improved precision of QTL mapping using a nonlinear Bayesian method in a multi-breed population leads to greater accuracy of across-breed genomic predictions. Genet. Sel. Evol. 2015, 47, 29. [Google Scholar] [CrossRef] [Green Version]
- Evangelou, E.; Ioannidis, J.P.A. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 2013, 14, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Leroy, G. Inbreeding depression in livestock species: Review and meta-analysis. Anim. Genet. 2014, 45, 618–628. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, I.; Boichard, D.; Lund, M.S. Comparing power and precision of within-breed and multibreed genome-wide association studies of production traits using whole-genome sequence data for 5 French and Danish dairy cattle breeds. J. Dairy Sci. 2016, 99, 8932–8945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, H.; Rosado, P.; Roser, M. Meat and Dairy Production. Our World in Data. 2017. Available online: https://ourworldindata.org/meat-production (accessed on 8 February 2023).
- Ponzoni, R.W.; Davies, R.L. An evaluation of biological and conventional pig selection indices. Aust. J. Exp. Agric. 1989, 29, 775–779. [Google Scholar] [CrossRef]
- McKay, R.M. Responses to index selection for reduced backfat thickness and increased growth rate in swine. Can. J. Anim. Sci. 1990, 70, 973–977. [Google Scholar] [CrossRef]
- Martínez-Montes, Á.M.; Fernández, A.; Muñoz, M.; Noguera, J.L.; Folch, J.M.; Fernández, A.I. Using genome wide association studies to identify common QTL regions in three different genetic backgrounds based on Iberian pig breed. PLoS ONE 2018, 13, e0190184. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Qiu, H.; Xiao, S.; Wu, Z.; Yang, M.; Yang, J.; Ren, J.; Huang, L. A genome-wide association study identifies genomic loci associated with backfat thickness, carcass weight, and body weight in two commercial pig populations. J. Appl. Genet. 2017, 58, 499–508. [Google Scholar] [CrossRef]
- Zhou, S.; Ding, R.; Meng, F.; Wang, X.; Zhuang, Z.; Quan, J.; Geng, Q.; Wu, J.; Zheng, E.; Wu, Z.; et al. A meta-analysis of genome-wide association studies for average daily gain and lean meat percentage in two Duroc pig populations. BMC Genom. 2021, 22, 12. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Bland, J.M.; Altman, D.G. Multiple significance tests: The Bonferroni method. BMJ 1995, 310, 170. [Google Scholar] [CrossRef] [Green Version]
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef] [Green Version]
- Verbyla, K.L.; Hayes, B.J.; Bowman, P.J.; Goddard, M.E. Accuracy of genomic selection using stochastic search variable selection in Australian Holstein Friesian dairy cattle. Genet. Res. 2009, 91, 307–311. [Google Scholar] [CrossRef]
- Misztal, I.; Tsuruta, S.; Lourenco, D.; Masuda, Y.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 Family Programs; University of Georgia: Athens, Greek, 2018. [Google Scholar]
- Masuda, Y. Introduction to BLUPF90 Suite Programs; University of Georgia: Athens, Greek, 2018. [Google Scholar]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Gao, N.; Martini, J.W.R.; Simianer, H. Integrating Gene Expression Data Into Genomic Prediction. Front. Genet. 2019, 10, 126. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.K.; Jeong, Y.D.; Cho, E.S.; Choi, T.J.; Kim, Y.M.; Cho, K.H.; Lee, J.B.; Lim, H.T.; Lee, D.H. A genome-wide association study of social genetic effects in Landrace pigs. Asian-Australas. J. Anim. Sci. 2018, 31, 784–790. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, B.; Yang, M.; Han, H.; Chen, T.; Wei, Q.; Miao, Z.; Yin, L.; Wang, R.; Shen, J.; et al. Genome-Wide Association Study and Fine Mapping Reveals Candidate Genes for Birth Weight of Yorkshire and Landrace Pigs. Front. Genet. 2020, 11, 183. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Xing, Y.; Ren, L.; Wang, Y.; Li, Q.; Yang, Q.; Du, M.; Xu, L.; Willems, L.; Li, J.; et al. bta-miR-23a Regulates the Myogenic Differentiation of Fetal Bovine Skeletal Muscle-Derived Progenitor Cells by Targeting MDFIC Gene. Genes 2020, 11, 1232. [Google Scholar] [CrossRef]
- Zhang, L.; Xiang, Z.; Michal, J.J.; Ding, B.; Rui, L.; Jiang, Z. Genome Wide Screening of Candidate Genes for Improving Piglet Birth Weight Using High and Low Estimated Breeding Value Populations. Int. J. Biol. Sci. 2014, 10, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Falker-Gieske, C.; Blaj, I.; Preuß, S.; Bennewitz, J.; Thaller, G.; Tetens, J. GWAS for Meat and Carcass Traits Using Imputed Sequence Level Genotypes in Pooled F2-Designs in Pigs. G3 Genes|Genomes|Genet. 2019, 9, 2823–2834. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.; Kiparaki, M.; Folgado, V.; Kumar, A.; Blanco, J.; Rimesso, G.; Chuen, J.; Liu, Y.; Zheng, D.; Baker, N.E. Drosophila RpS12 controls translation, growth, and cell competition through Xrp1. PLoS Genet. 2019, 15, e1008513. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.L.C.; Richard, F.J.; Kuo, W.-P.; D’Ercole, A.J.; Conti, M. Impaired growth and fertility of cAMP-specific phosphodiesterase PDE4D-deficient mice. Proc. Natl. Acad. Sci. USA 1999, 96, 11998–12003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancar, G.; Liu, S.; Gasser, E.; Alvarez, J.G.; Moutos, C.; Kim, K.; van Zutphen, T.; Wang, Y.; Huddy, T.F.; Ross, B.; et al. FGF1 and insulin control lipolysis by convergent pathways. Cell Metab. 2022, 34, 171–183.e6. [Google Scholar] [CrossRef]
- Jiang, L.; Li, J.; Liu, X.; Burnstock, G.; Xiang, Z. Expression of aquaporin-4 water channels in the digestive tract of the guinea pig. J. Mol. Histol. 2014, 45, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.S.; Komar, A.R.; Ma, T.; Filiz, F.; McLeroy, J.; Hoda, K.; Verkman, A.S.; Bastidas, J.A. Gastric acid secretion in aquaporin-4 knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G448–G453. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Goldberg, E.B.; Makarova, K.S.; Lin, L.; Brown, W.J.; Jackson, C.L. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep 2006, 7, 106–113. [Google Scholar] [CrossRef]
- Crespo-Piazuelo, D.; Criado-Mesas, L.; Revilla, M.; Castelló, A.; Noguera, J.L.; Fernández, A.I.; Ballester, M.; Folch, J.M. Identification of strong candidate genes for backfat and intramuscular fatty acid composition in three crosses based on the Iberian pig. Sci. Rep. 2020, 10, 13962. [Google Scholar] [CrossRef]
- Yip, L.; Fathman, C.G. Type 1 diabetes in mice and men: Gene expression profiling to investigate disease pathogenesis. Immunol. Res. 2014, 58, 340–350. [Google Scholar] [CrossRef]
- Lukić, B.; Ferenčaković, M.; Šalamon, D.; Čačić, M.; Orehovački, V.; Iacolina, L.; Curik, I.; Cubric-Curik, V. Conservation Genomic Analysis of the Croatian Indigenous Black Slavonian and Turopolje Pig Breeds. Front. Genet. 2020, 11, 261. [Google Scholar] [CrossRef]
- Nakamura, Y.; Kanemarum, K.; Fukami, K. Physiological functions of phospholipase Cδ1 and phospholipase Cδ3. Adv. Biol. Regul. 2013, 53, 356–362. [Google Scholar] [CrossRef]
- Qiao, R.; Zhang, M.; Zhang, B.; Li, X.; Han, X.; Wang, K.; Li, X.; Yang, F.; Hu, P. Population genetic structure analysis and identification of backfat thickness loci of Chinese synthetic Yunan pigs. Front. Genet. 2022, 13, 1039838. [Google Scholar] [CrossRef]
- Taye, M.; Kim, J.; Yoon, S.H.; Lee, W.; Hanotte, O.; Dessie, T.; Kemp, S.; Mwai, O.A.; Caetano-Anolles, K.; Cho, S.; et al. Whole genome scan reveals the genetic signature of African Ankole cattle breed and potential for higher quality beef. BMC Genet. 2017, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Li-Gao, R.; Hughes, D.A.; van Klinken, J.B.; de Mutsert, R.; Rosendaal, F.R.; Mook-Kanamori, D.O.; Timpson, N.J.; Willems van Dijk, K. Genetic Studies of Metabolomics Change After a Liquid Meal Illuminate Novel Pathways for Glucose and Lipid Metabolism. Diabetes 2021, 70, 2932–2946. [Google Scholar] [CrossRef] [PubMed]
- Harder, M.N.; Ribel-Madsen, R.; Justesen, J.M.; Sparsø, T.; Andersson, E.A.; Grarup, N.; Jørgensen, T.; Linneberg, A.; Hansen, T.; Pedersen, O. Type 2 Diabetes Risk Alleles Near BCAR1 and in ANK1 Associate With Decreased β-Cell Function Whereas Risk Alleles Near ANKRD55 and GRB14 Associate With Decreased Insulin Sensitivity in the Danish Inter99 Cohort. J. Clin. Endocrinol. Metab. 2013, 98, E801–E806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taneera, J.; Mohammed, A.K.; Dhaiban, S.; Hamad, M.; Prasad, R.B.; Sulaiman, N.; Salehi, A. RORB and RORC associate with human islet dysfunction and inhibit insulin secretion in INS-1 cells. Islets 2019, 11, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roforth, M.M.; Liu, G.; Khosla, S.; Monroe, D.G. Examination of nuclear receptor expression in osteoblasts reveals Rorβ as an important regulator of osteogenesis. J. Bone Miner. Res. 2012, 27, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Hovenier, R.; Kanis, E.; van Asseldonk, T.; Westerink, N.G. Genetic parameters of pig meat quality traits in a halothane negative population. Livest. Prod. Sci. 1992, 32, 309–321. [Google Scholar] [CrossRef]
- Suzuki, K.; Kadowaki, H.; Shibata, T.; Uchida, H.; Nishida, A. Selection for daily gain, loin-eye area, backfat thickness and intramuscular fat based on desired gains over seven generations of Duroc pigs. Livest. Prod. Sci. 2005, 97, 193–202. [Google Scholar] [CrossRef]
- Suzuki, K.; Irie, M.; Kadowaki, H.; Shibata, T.; Kumagai, M.; Nishida, A. Genetic parameter estimates of meat quality traits in Duroc pigs selected for average daily gain, longissimus muscle area, backfat thickness, and intramuscular fat content. J. Anim. Sci. 2005, 83, 2058–2065. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Zeng, Y.; Wang, H.; Yang, L.; Yang, Y.; Zhu, H.; Shi, Y.; Chen, W.; Hu, Y. Molecular characterization and expression analysis of NDUFS4 gene in m. longissimus dorsi of Laiwu pig (Sus scrofa). Mol. Biol. Rep. 2013, 40, 1599–1608. [Google Scholar] [CrossRef]
- Grapes, L.; Rothschild, M.F. Investigation of a QTL region for loin eye area and fatness on pig Chromosome 1. Mamm. Genome 2006, 17, 657–668. [Google Scholar] [CrossRef]
- Schneider, J.F.; Rempel, L.A.; Snelling, W.M.; Wiedmann, R.T.; Nonneman, D.J.; Rohrer, G.A. Genome-wide association study of swine farrowing traits. Part II: Bayesian analysis of marker data. J. Anim. Sci. 2012, 90, 3360–3367. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, D.; Chen, X.; Hu, M.; Wang, J.a.; Li, Y.; Guo, N.; Shen, B. cDNA cloning and function analysis of two novel erythroid differentiation related genes. Sci. China Ser. C Life Sci. 2001, 44, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Alli, Z.; Ackerley, C.; Chen, Y.; Al-Saud, B.; Abdelhaleem, M. Nuclear and mitochondrial localization of the putative RNA helicase DHX32. Exp. Mol. Pathol. 2006, 81, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Kroll, K.L. Geminin in embryonic development: Coordinating transcription and the cell cycle during differentiation. FBL 2007, 12, 1395–1409. [Google Scholar] [CrossRef] [Green Version]
- Oatley, J.M.; Brinster, R.L. Regulation of spermatogonial stem cell self-renewal in mammals. Annu. Rev. Cell Dev. Biol. 2008, 24, 263–286. [Google Scholar] [CrossRef] [Green Version]
- Stephens, M.; Balding, D.J. Bayesian statistical methods for genetic association studies. Nat. Rev. Genet. 2009, 10, 681–690. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef] [Green Version]
- Alpay, F.; Zare, Y.; Kamalludin, M.H.; Huang, X.; Shi, X.; Shook, G.; Collins, M.; Kirkpatrick, B. Genome-Wide Association Study of Susceptibility to Infection by Mycobacterium avium Subspecies paratuberculosis in Holstein Cattle. PLoS ONE 2014, 9, e111704. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Su, G.; Christensen, O.F.; Janss, L.; Lund, M.S. Genome-wide association analyses using a Bayesian approach for litter size and piglet mortality in Danish Landrace and Yorkshire pigs. BMC Genom. 2016, 17, 468. [Google Scholar] [CrossRef] [Green Version]
- Zare, Y.; Shook, G.E.; Collins, M.T.; Kirkpatrick, B.W. Genome-Wide Association Analysis and Genomic Prediction of Mycobacterium avium Subspecies paratuberculosis Infection in US Jersey Cattle. PLoS ONE 2014, 9, e88380. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Hickey, J.M.; Cleveland, M.A.; Maltecca, C. Assessment of alternative genotyping strategies to maximize imputation accuracy at minimal cost. Genet. Sel. Evol. 2012, 44, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esfandyari, H.; Sørensen, A.C.; Bijma, P. A crossbred reference population can improve the response to genomic selection for crossbred performance. Genet. Sel. Evol. 2015, 47, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Clark, S.; van der Werf, J.H.J. Estimation of genomic prediction accuracy from reference populations with varying degrees of relationship. PLoS ONE 2017, 12, e0189775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.; Fernandez, M.M.; Steen, H.; Blagoev, B.; Nielsen, M.M.; Roche, S.; Mann, M.; Lodish, H.F. Identification of a Novel Immunoreceptor Tyrosine-based Activation Motif-containing Molecule, STAM2, by Mass Spectrometry and Its Involvement in Growth Factor and Cytokine Receptor Signaling Pathways. J. Biol. Chem. 2000, 275, 38633–38639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.Q.; Hui, Y.T.; Liu, R.Y.; Jiao, R.G.; Gong, Y.; Zhang, Y.Y.; Xie, H.Q. Molecular cloning, polymorphisms, and association analysis of the promoter region of the STAM2 gene in Wuchuan Black cattle. Genet. Mol. Res. 2013, 12, 3651–3661. [Google Scholar] [CrossRef]
- Mead, T.J.; Martin, D.R.; Wang, L.W.; Cain, S.A.; Gulec, C.; Cahill, E.; Mauch, J.; Reinhardt, D.P.; Lo, C.W.; Baldock, C.; et al. Proteolysis of fibrillin-2 microfibrils is essential for normal skeletal development. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Kearns, A.E.; Donohue, M.M.; Sanyal, B.; Demay, M.B. Cloning and Characterization of a Novel Protein Kinase That Impairs Osteoblast Differentiation in Vitro. J. Biol. Chem. 2001, 276, 42213–42218. [Google Scholar] [CrossRef] [Green Version]
- Skiba, G.; Sobol, M.; Raj, S. Bone mineralization, geometry and strength in pigs growing from 56 to 115 day of life as affected by body fatness. J. Anim. Feed Sci. 2016, 25, 302–308. [Google Scholar] [CrossRef]
- Pérez-Enciso, M.; Forneris, N.; de los Campos, G.; Legarra, A. Evaluating Sequence-Based Genomic Prediction with an Efficient New Simulator. Genetics 2017, 205, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Ye, S.; Jiang, Y.; Zhang, Z.; Zhang, Q.; Ding, X. Using imputation-based whole-genome sequencing data to improve the accuracy of genomic prediction for combined populations in pigs. Genet. Sel. Evol. 2019, 51, 58. [Google Scholar] [CrossRef]
- Calus, M.P.L.; Bouwman, A.C.; Schrooten, C.; Veerkamp, R.F. Efficient genomic prediction based on whole-genome sequence data using split-and-merge Bayesian variable selection. Genet. Sel. Evol. 2016, 48, 49. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, I.; Boichard, D.; Guldbrandtsen, B.; Lund, M.S. Using Sequence Variants in Linkage Disequilibrium with Causative Mutations to Improve Across-Breed Prediction in Dairy Cattle: A Simulation Study. G3 Genes|Genomes|Genet. 2016, 6, 2553–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veerkamp, R.F.; Bouwman, A.C.; Schrooten, C.; Calus, M.P.L. Genomic prediction using preselected DNA variants from a GWAS with whole-genome sequence data in Holstein–Friesian cattle. Genet. Sel. Evol. 2016, 48, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.; Gao, N.; Zheng, R.; Chen, Z.; Teng, J.; Yuan, X.; Zhang, H.; Chen, Z.; Zhang, X.; Li, J.; et al. Strategies for Obtaining and Pruning Imputed Whole-Genome Sequence Data for Genomic Prediction. Front. Genet. 2019, 10, 673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ober, U.; Ayroles, J.F.; Stone, E.A.; Richards, S.; Zhu, D.; Gibbs, R.A.; Stricker, C.; Gianola, D.; Schlather, M.; Mackay, T.F.C.; et al. Using Whole-Genome Sequence Data to Predict Quantitative Trait Phenotypes in Drosophila melanogaster. PLoS Genet. 2012, 8, e1002685. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Trait | N | Mean ± SD | CV | Max | Min | h2 |
|---|---|---|---|---|---|---|---|
| Yorkshire | ADG | 1734 | 565.32 ± 44.92 | 7.95 | 792.59 | 394.51 | 0.44 |
| BFT | 1734 | 11.09 ± 3.34 | 30.12 | 28.04 | 5.17 | 0.43 | |
| BTHWT | 887 | 1.39 ± 0.25 | 17.99 | 2.42 | 0.60 | 0.16 | |
| BW | 1734 | 112.45 ± 14.64 | 13.02 | 167.10 | 74.01 | 0.42 | |
| Landrace | ADG | 1123 | 608.19 ± 53.69 | 8.83 | 856.78 | 421.54 | 0.44 |
| BFT | 1123 | 14.58 ± 4.88 | 33.47 | 34.95 | 5.46 | 0.40 | |
| BTHWT | 405 | 1.42 ± 0.26 | 18.31 | 2.17 | 0.71 | 0.27 | |
| BW | 1123 | 117.12 ± 16.25 | 13.87 | 160.12 | 80.09 | 0.38 |
| Traits 1 | Breeds 2 | The Number of Significant SNPs | Candidate Genes | ||
|---|---|---|---|---|---|
| CL_GWAS 3 | CB_GWAS 4 | IL_GWAS 5 | |||
| ADG | YY | 14 | 242 (12) | 6 | MDFIC, FOXP2, DOCK4, IMMP2L, ZPLD1, CYP7B1 |
| LL | 3 | 164 (7) | 1 | ALDH8A1, RPS12 | |
| BFT | YY | 20 | 219 (11) | 71 | UMAD1, GLCCI1, PDE4D, ZSWIM6, RNF180, ANKRD55, NDUFS4, NDUFA4 |
| LL | 5 | 193 (12) | 2 | ODF3, DEAF1, PACS1, ZNF300, MS4A8, MS4A13 | |
| BTHWT | YY | 4 | 20 | EDRF1, DHX32, GMNN, MPP7, CUBN, ITGA8, RPP38, UCMA | |
| LL | 104 (5) | ASAP1, NAV3, MROH5, PTP4A3, GPR20 | |||
| BW | YY | 6 | 245 (12) | 40 | TAF4B, AQP4, RORB, ATXN1, TAFA5, SELENOI, TMEM104 |
| LL | 1 | 174 (10) | 2 | AMER2, MTMR6, NUP58, ATP8A2, SHISA2, FAM171A1 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Zhang, Y.; Liu, T.; Jiang, B.; Li, Z.; Qu, Y.; Chen, Y.; Li, Z. Utilizing Variants Identified with Multiple Genome-Wide Association Study Methods Optimizes Genomic Selection for Growth Traits in Pigs. Animals 2023, 13, 722. https://doi.org/10.3390/ani13040722
Zhang R, Zhang Y, Liu T, Jiang B, Li Z, Qu Y, Chen Y, Li Z. Utilizing Variants Identified with Multiple Genome-Wide Association Study Methods Optimizes Genomic Selection for Growth Traits in Pigs. Animals. 2023; 13(4):722. https://doi.org/10.3390/ani13040722
Chicago/Turabian StyleZhang, Ruifeng, Yi Zhang, Tongni Liu, Bo Jiang, Zhenyang Li, Youping Qu, Yaosheng Chen, and Zhengcao Li. 2023. "Utilizing Variants Identified with Multiple Genome-Wide Association Study Methods Optimizes Genomic Selection for Growth Traits in Pigs" Animals 13, no. 4: 722. https://doi.org/10.3390/ani13040722