
Integrative Analysis of Blood Transcriptomics and Metabolomics Reveals Molecular Regulation of Backfat Thickness in Qinchuan Cattle

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Phenotypes
2.2. Blood Sample Collection and Preparation
2.3. RNA Extraction, Sequencing, and Transcriptome Data Analysis
2.4. Metabolites Extraction
2.5. LC-MS/MS Analysis
2.6. Joint Analysis of the Transcriptomic and Metabolomic Data
2.7. Statistical Analyses
3. Results
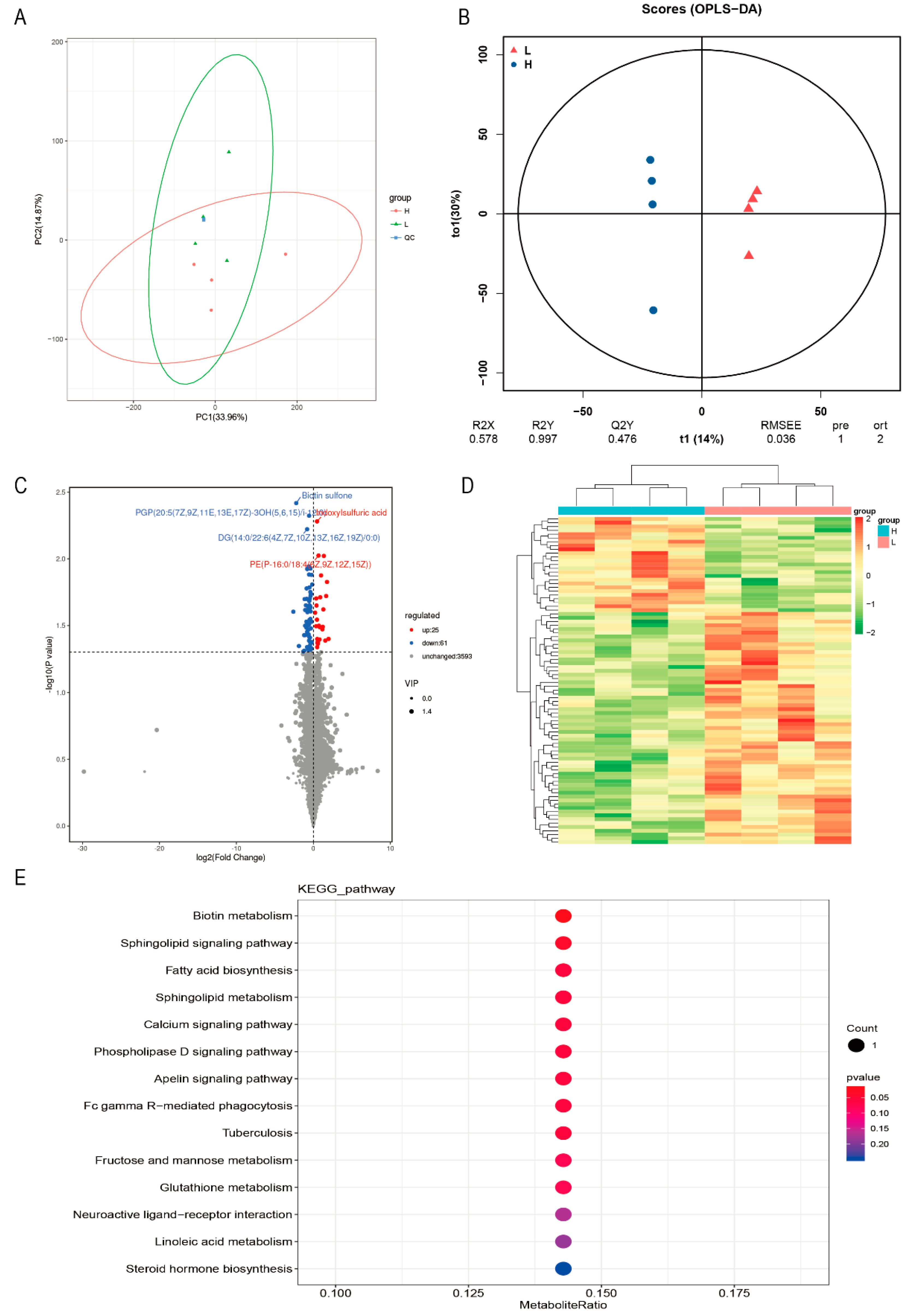
3.1. Animal Phenotypic Divergence for BFT
3.2. DEGs and Transcriptome Analysis
3.3. DEMs and Metabolome Analysis
3.4. Joint Analysis of the Transcriptome and Metabolome
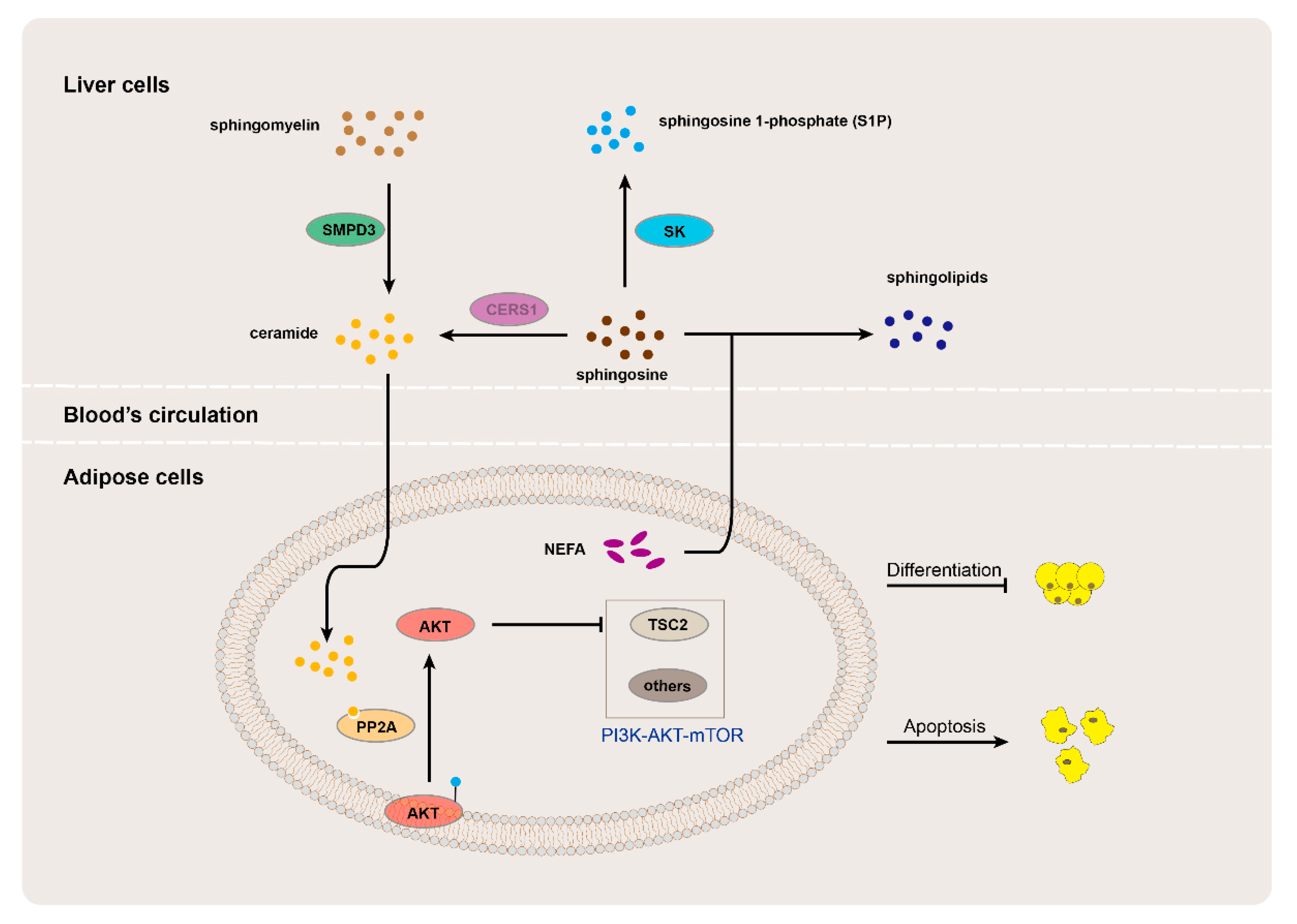
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berry, D.P.; Conroy, S.; Pabiou, T.; Cromie, A.R. Animal breeding strategies can improve meat quality attributes within entire populations. Meat Sci. 2017, 132, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Polkinghorne, R.J.; Thompson, J.M. Meat standards and grading: A world view. Meat Sci. 2010, 86, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Gol, S.; González-Prendes, R.; Bosch, L.; Tor, M.; Reixach, J.; Pena, R.N.; Estany, J. Linoleic acid metabolic pathway allows for an efficient increase of intramuscular fat content in pigs. J. Anim. Sci. Biotechnol. 2019, 10, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merks, J.W.M.; Mathur, P.K.; Knol, E.F. New phenotypes for new breeding goals in pigs. Animal 2012, 6, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.B.; Crouse, J.D. Relative contributions of acetate, lactate and glucose to lipogenesis in bovine intramuscular and subcutaneous adipose tissue. J. Nutr. 1984, 114, 792–800. [Google Scholar] [CrossRef]
- Dou, T.; Yan, S.; Liu, L.; Wang, K.; Jian, Z.; Xu, Z.; Zhao, J.; Wang, Q.; Sun, S.; Talpur, M.Z.; et al. Integrative analysis of transcriptomics and metabolomics to reveal the melanogenesis pathway of muscle and related meat characters in Wuliangshan black-boned chickens. BMC Genom. 2022, 23, 173. [Google Scholar] [CrossRef]
- Zhan, H.; Xiong, Y.; Wang, Z.; Dong, W.; Zhou, Q.; Xie, S.; Li, X.; Zhao, S.; Ma, Y. Integrative analysis of transcriptomic and metabolomic profiles reveal the complex molecular regulatory network of meat quality in Enshi black pigs. Meat Sci. 2022, 183, 108642. [Google Scholar] [CrossRef]
- Magalhães, A.F.B.; Schenkel, F.S.; Garcia, D.A.; Gordo, D.G.M.; Tonussi, R.L.; Espigolan, R.; Silva, R.M.d.O.; Braz, C.U.; Fernandes Júnior, G.A.; Baldi, F.; et al. Genomic selection for meat quality traits in Nelore cattle. Meat Sci. 2019, 148, 32–37. [Google Scholar] [CrossRef]
- Maciel, F.C.; Machado Neto, O.R.; Duarte, M.S.; Du, M.; Lage, J.F.; Teixeira, P.D.; Martins, C.L.; Domingues, E.H.R.; Fogaça, L.A.; Ladeira, M.M. Effect of vitamin A injection at birth on intramuscular fat development and meat quality in beef cattle. Meat Sci. 2022, 184, 108676. [Google Scholar] [CrossRef]
- Iannaccone, M.; Ianni, A.; Contaldi, F.; Esposito, S.; Martino, C.; Bennato, F.; De Angelis, E.; Grotta, L.; Pomilio, F.; Giansante, D.; et al. Whole blood transcriptome analysis in ewes fed with hemp seed supplemented diet. Sci. Rep. 2019, 9, 16192. [Google Scholar] [CrossRef] [Green Version]
- Connolly, S.; Dona, A.; Wilkinson-White, L.; Hamblin, D.; D’Occhio, M.; González, L.A. Relationship of the blood metabolome to subsequent carcass traits at slaughter in feedlot Wagyu crossbred steers. Sci. Rep. 2019, 9, 15139. [Google Scholar] [CrossRef] [Green Version]
- Wojciechowicz, B.; Kołakowska, J.; Zglejc-Waszak, K.; Martyniak, M.; Kotwica, G.; Franczak, A. The whole blood transcriptome at the time of maternal recognition of pregnancy in pigs reflects certain alterations in gene expression within the endometrium and the myometrium. Theriogenology 2019, 126, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhang, Y.; Qin, L.; Ma, W.; Zhu, L.; Luo, X. Association of ultimate pH and stress-related blood variables in cattle. Meat Sci. 2018, 139, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H. The perspective of meat and meat-alternative consumption in China. Meat Sci. 2022, 194, 108982. [Google Scholar] [CrossRef] [PubMed]
- Hengwei, Y.; Raza, S.H.A.; Wang, S.; Khan, R.; Ayari-Akkari, A.; El Moneim Ahmed, D.A.; Ahmad, I.; Shaoib, M.; Abd El-Aziz, A.H.; Rahman, S.U.; et al. The growth curve determination and economic trait correlation for Qinchuan bull population. Anim. Biotechnol. 2022, 1–8. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Chen, L.; Tian, S.; Lin, Y.; Tang, Q.; Zhou, X.; Li, D.; Yeung, C.K.L.; Che, T.; Jin, L.; et al. Comprehensive variation discovery and recovery of missing sequence in the pig genome using multiple de novo assemblies. Genome Res. 2017, 27, 865–874. [Google Scholar] [CrossRef] [Green Version]
- Noy, N. The one-two punch: Retinoic acid suppresses obesity both by promoting energy expenditure and by inhibiting adipogenesis. Adipocyte 2013, 2, 184–187. [Google Scholar] [CrossRef] [Green Version]
- Quenneville, S.; Verde, G.; Corsinotti, A.; Kapopoulou, A.; Jakobsson, J.; Offner, S.; Baglivo, I.; Pedone, P.V.; Grimaldi, G.; Riccio, A.; et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell 2011, 44, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Bina, M. Imprinted control regions include composite DNA elements consisting of the ZFP57 binding site overlapping MLL1 morphemes. Genomics 2017, 109, 265–273. [Google Scholar] [CrossRef]
- Volkov, P.; Olsson, A.H.; Gillberg, L.; Jørgensen, S.W.; Brøns, C.; Eriksson, K.-F.; Groop, L.; Jansson, P.-A.; Nilsson, E.; Rönn, T.; et al. A Genome-Wide mQTL Analysis in Human Adipose Tissue Identifies Genetic Variants Associated with DNA Methylation, Gene Expression and Metabolic Traits. PLoS ONE 2016, 11, e0157776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, D.J.G.; Callaway, J.L.A.; Marks, S.M.; White, H.E.; Acerini, C.L.; Boonen, S.E.; Dayanikli, P.; Firth, H.V.; Goodship, J.A.; Haemers, A.P.; et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat. Genet. 2008, 40, 949–951. [Google Scholar] [CrossRef]
- Fang, Z.; Pyne, S.; Pyne, N.J. Ceramide and sphingosine 1-phosphate in adipose dysfunction. Prog. Lipid Res. 2019, 74, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Meikle, P.J.; Summers, S.A. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nat. Rev. Endocrinol. 2017, 13, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Yiew, N.K.H.; Chatterjee, T.K.; Tang, Y.L.; Pellenberg, R.; Stansfield, B.K.; Bagi, Z.; Fulton, D.J.; Stepp, D.W.; Chen, W.; Patel, V.; et al. A novel role for the Wnt inhibitor APCDD1 in adipocyte differentiation: Implications for diet-induced obesity. J. Biol. Chem. 2017, 292, 6312–6324. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.G.; Trevaskis, J.L.; Lam, D.D.; Sutton, G.M.; Koza, R.A.; Chouljenko, V.N.; Kousoulas, K.G.; Rogers, P.M.; Kesterson, R.A.; Thearle, M.; et al. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 2008, 8, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Qiu, B.; Lee, D.S.M.; Walton, Z.E.; Ochocki, J.D.; Mathew, L.K.; Mancuso, A.; Gade, T.P.F.; Keith, B.; Nissim, I.; et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 2014, 513, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Venegas-Calerón, M.; Sayanova, O.; Napier, J.A. An alternative to fish oils: Metabolic engineering of oil-seed crops to produce omega-3 long chain polyunsaturated fatty acids. Prog. Lipid Res. 2010, 49, 108–119. [Google Scholar] [CrossRef]
- Smaldone, G.; Pirone, L.; Capolupo, A.; Vitagliano, L.; Monti, M.C.; Di Gaetano, S.; Pedone, E. The essential player in adipogenesis GRP78 is a novel KCTD15 interactor. Int. J. Biol. Macromol. 2018, 115, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Aburasayn, H.; Al Batran, R.; Ussher, J.R. Targeting ceramide metabolism in obesity. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E423–E435. [Google Scholar] [CrossRef] [Green Version]
- Chaurasia, B.; Summers, S.A. Ceramides—Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Blouin, C.M.; Prado, C.; Takane, K.K.; Lasnier, F.; Garcia-Ocana, A.; Ferré, P.; Dugail, I.; Hajduch, E. Plasma membrane subdomain compartmentalization contributes to distinct mechanisms of ceramide action on insulin signaling. Diabetes 2010, 59, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolak, M.; Westerbacka, J.; Velagapudi, V.R.; Wågsäter, D.; Yetukuri, L.; Makkonen, J.; Rissanen, A.; Häkkinen, A.-M.; Lindell, M.; Bergholm, R.; et al. Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes 2007, 56, 1960–1968. [Google Scholar] [CrossRef] [Green Version]
- Pyne, S.; Chapman, J.; Steele, L.; Pyne, N.J. Sphingomyelin-derived lipids differentially regulate the extracellular signal-regulated kinase 2 (ERK-2) and c-Jun N-terminal kinase (JNK) signal cascades in airway smooth muscle. Eur. J. Biochem. 1996, 237, 819–826. [Google Scholar] [CrossRef]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef]
- Bellini, L.; Campana, M.; Mahfouz, R.; Carlier, A.; Véret, J.; Magnan, C.; Hajduch, E.; Le Stunff, H. Targeting sphingolipid metabolism in the treatment of obesity/type 2 diabetes. Expert Opin. Ther. Targets 2015, 19, 1037–1050. [Google Scholar] [CrossRef]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Raza, S.H.A.; Pan, Y.; Cheng, G.; Mei, C.; Zan, L. Integrative Analysis of Blood Transcriptomics and Metabolomics Reveals Molecular Regulation of Backfat Thickness in Qinchuan Cattle. Animals 2023, 13, 1060. https://doi.org/10.3390/ani13061060
Yu H, Raza SHA, Pan Y, Cheng G, Mei C, Zan L. Integrative Analysis of Blood Transcriptomics and Metabolomics Reveals Molecular Regulation of Backfat Thickness in Qinchuan Cattle. Animals. 2023; 13(6):1060. https://doi.org/10.3390/ani13061060
Chicago/Turabian StyleYu, Hengwei, Sayed Haidar Abbas Raza, Yueting Pan, Gong Cheng, Chugang Mei, and Linsen Zan. 2023. "Integrative Analysis of Blood Transcriptomics and Metabolomics Reveals Molecular Regulation of Backfat Thickness in Qinchuan Cattle" Animals 13, no. 6: 1060. https://doi.org/10.3390/ani13061060