
Alterations of the Gut Microbiota and Metabolomics Associated with the Different Growth Performances of Macrobrachium rosenbergii Families
 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cultivation Management
2.2. Experimental Groups
2.3. Sampling
2.4. 16S rRNA Sequencing
2.4.1. DNA Extraction, 16S rRNA Amplification, and Sequencing
2.4.2. Bioinformatics and Statistical Analysis of 16S rRNA Sequencing
2.5. Metabolomic Analyses
2.5.1. Identification of Metabolites
2.5.2. LC–MS Data Processing and Identification of Differential Metabolites
2.6. The Correlation Analysis between Gut Microbiota and Differential Metabolites
2.7. Statistical Analysis
3. Results
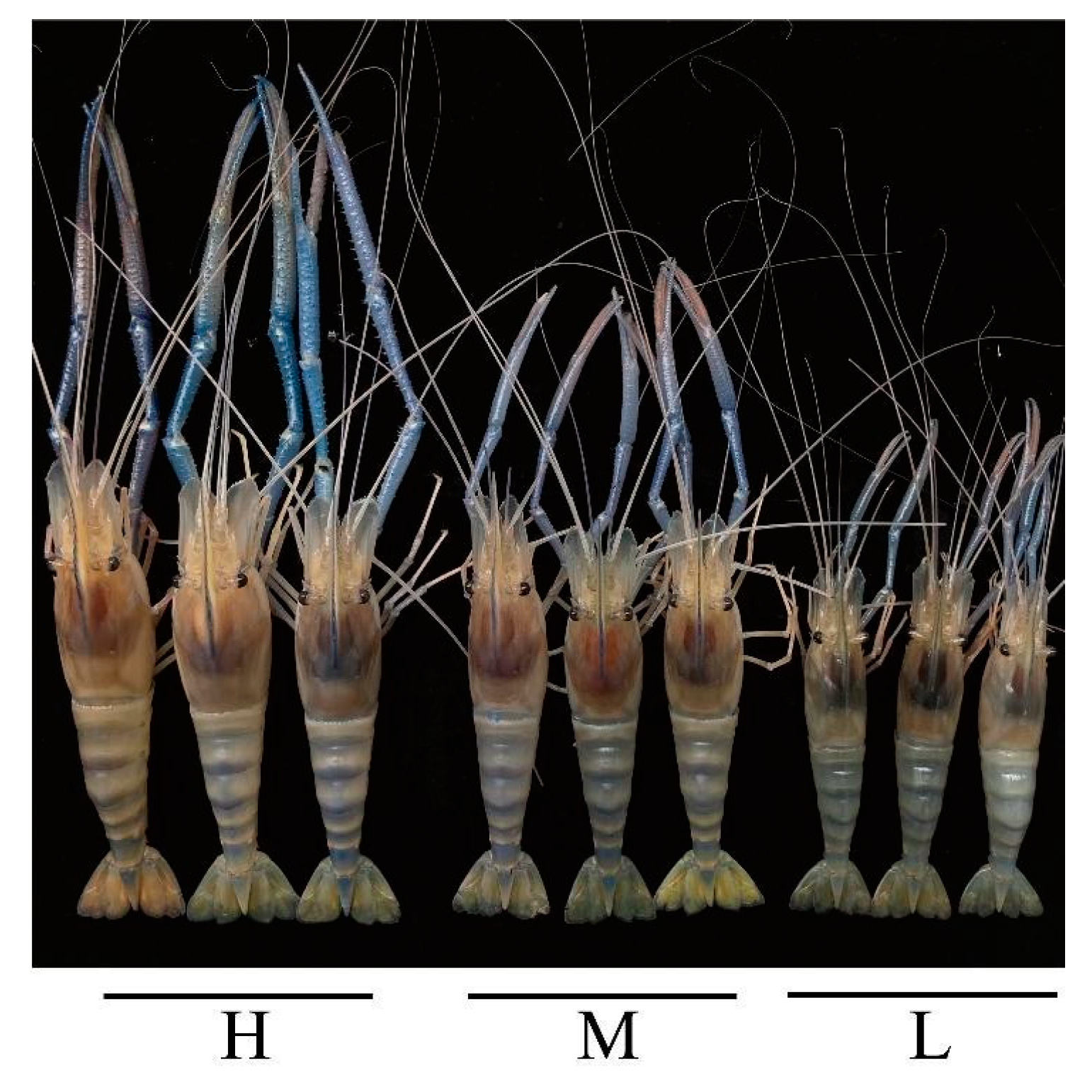
3.1. Growth Performance of GFP Families
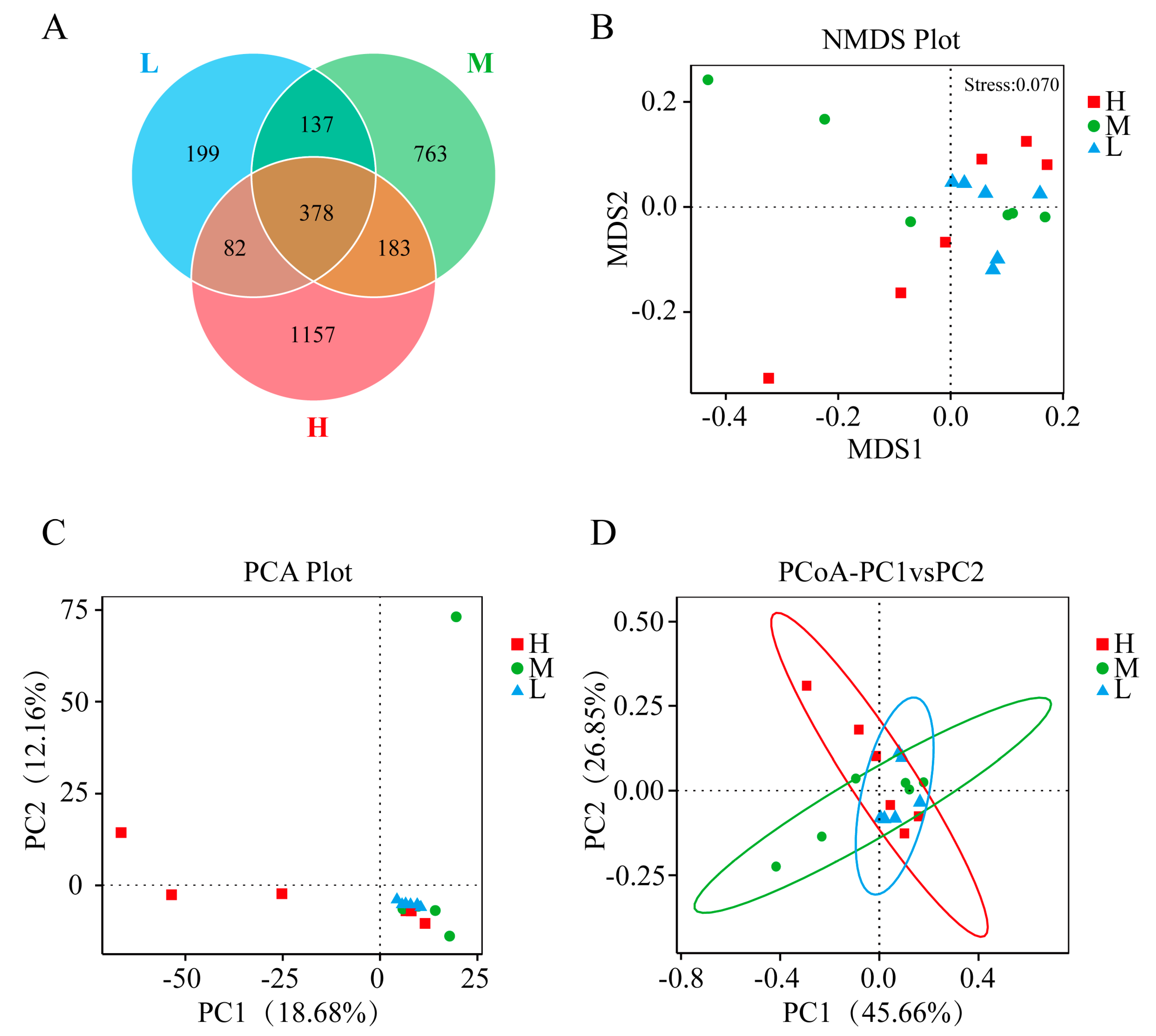
3.2. Gut Microbiota of GFP Families
3.2.1. Composition of the Gut Microbiota within the Three Levels of Growth Performance
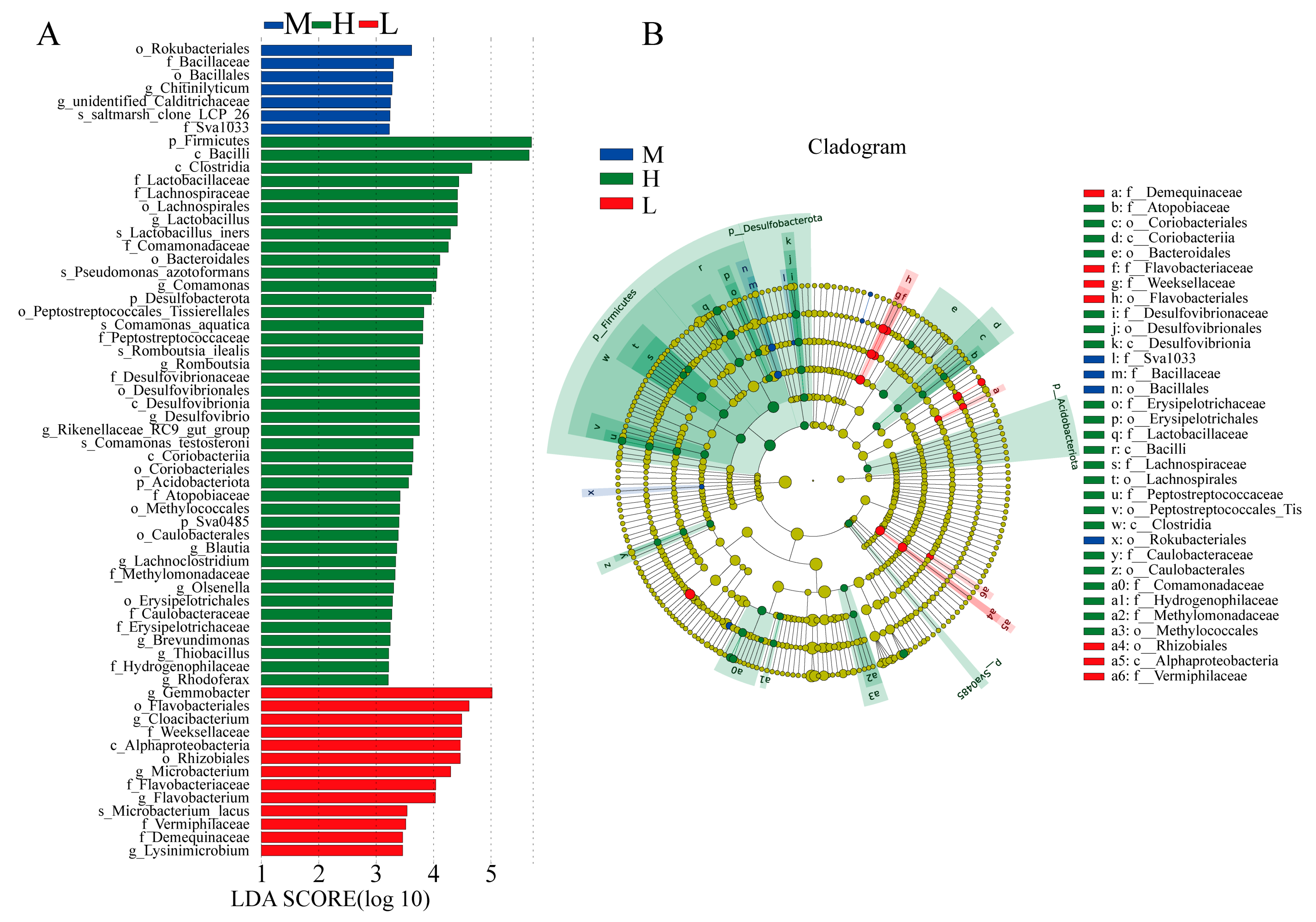
3.2.2. Identification of Differential Gut Microbiota within the Three Levels of Growth Performance
3.3. Metabolomics Analyses of GFP Families
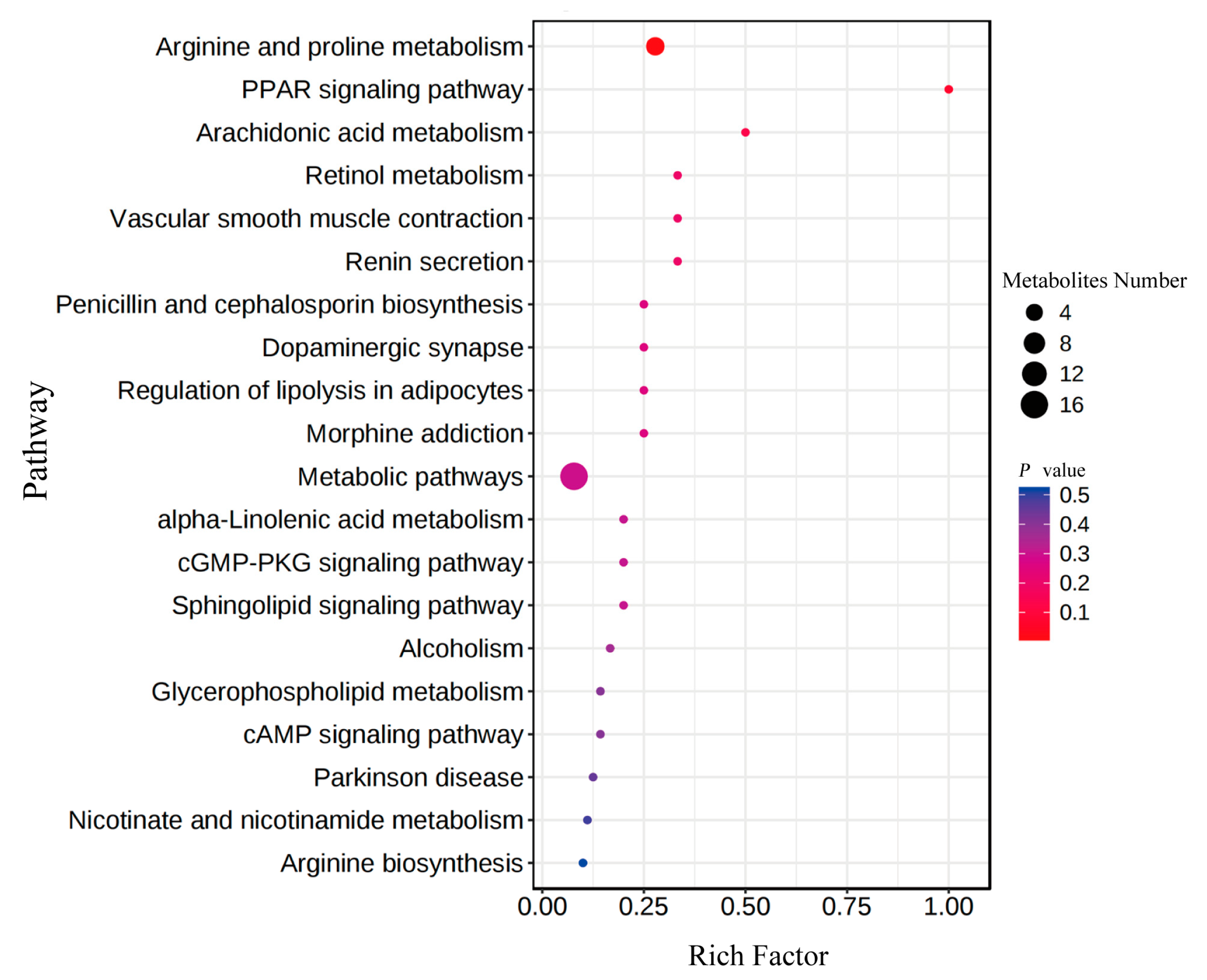
3.3.1. Identification of Differential Intestinal Metabolites among Three Groups
3.3.2. Key Roles of the Differential Metabolites in Growth Performance
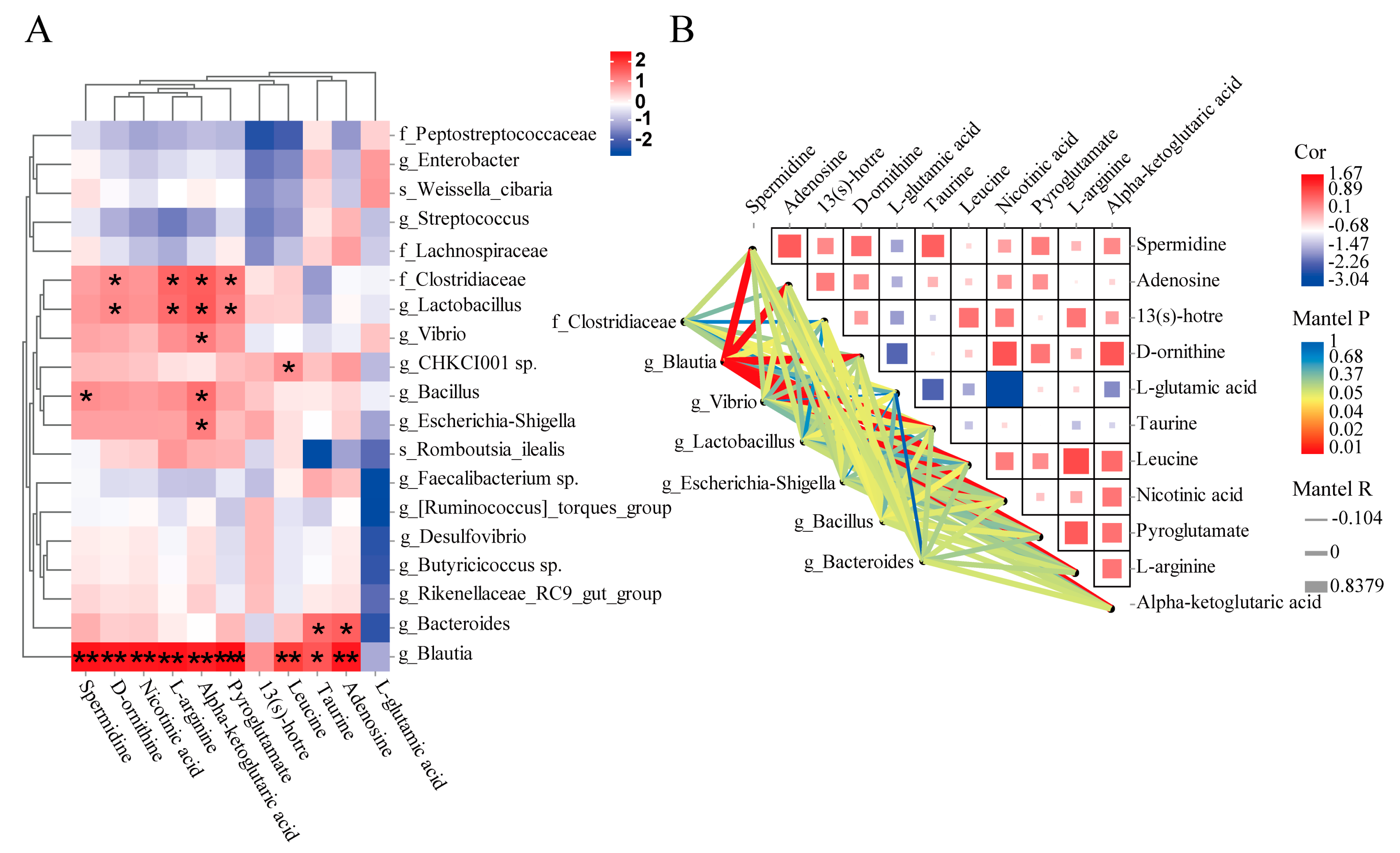
3.4. Correlation Analysis between Gut Microbiota and Metabolites
3.5. The Roles of the Metabolites Related to Metabolism of Key Amino Acids and Fatty Acids in Growth Performance of GFP
4. Discussion
4.1. Gut Microbiota Promotes the Growth and Metabolism of GFPs
4.2. Key Metabolites Play Important Roles in the Growth of GFPs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nguyen, M.T.; Raul, W.P.; Nguyen, H.N.; Nguyen, T.V.; Andrew, B.; Peter, B.M. Evaluation of growth performance in a diallel cross of three strains of giant freshwater prawn (Macrobrachium rosenbergii) in Vietnam. Aquaculture 2009, 287, 75–83. [Google Scholar] [CrossRef]
- Tan, K.; Li, Y.; Zhou, M.; Wang, W. siRNA knockdown of MrIR induces sex reversal in Macrobrachium rosenbergii. Aquaculture 2020, 523, 735172. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut Microbiota in Health and Disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.L.; Xu, Z.T. Significant Differences in Intestinal Microbial Communities in Aquatic Animals from an Aquaculture Area. J. Mar. Sci. Eng. 2021, 9, 104. [Google Scholar] [CrossRef]
- Xia, J.; Jin, C.; Pan, Z.; Sun, L.; Fu, Z.; Jin, Y. Chronic exposure to low concentrations of lead induces metabolic disorder and dysbiosis of the gut microbiota in mice. Sci. Total Environ. 2018, 631–632, 439–448. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Zmora, N.; Bashiardes, S.; Levy, M.; Elinav, E. The Role of the Immune System in Metabolic Health and Disease. Cell Metab. 2017, 25, 506–521. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, J. Role of intestinal microbiota and metabolites on gut homeostasis and human diseases. BMC Immunol. 2017, 18, 2. [Google Scholar] [CrossRef]
- Ringø, E.; Van, D.H.; Lee, S.H.; Soltani, M.; Hoseinifar, S.H.; Harikrishnan, R.; Song, S.K. Probiotics, lactic acid bacteria, and bacilli: Interesting supplementation for aquaculture. J. Appl. Microbiol. 2020, 129, 116–136. [Google Scholar] [CrossRef]
- Cardona, E.; Gueguen, Y.; Magré, K.; Lorgeoux, B.; Piquemal, D.; Pierrat, F.; Noguier, F.; Saulnier, D. Bacterial community characterization of water and intestine of the shrimp Litopenaeus stylirostris in a biofloc system. BMC Microbiol. 2016, 16, 157. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Dethlefsen, L.; McFall-Ngai, M.; Relman, D.A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 2007, 449, 811–818. [Google Scholar] [CrossRef]
- Rawls, J.F.; Mahowald, M.A.; Ley, R.E.; Gordon, J.I. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 2006, 127, 423–433. [Google Scholar] [CrossRef]
- Xiao, F.; Zhu, W.; Yu, Y.; He, Z.; Wu, B.; Wang, C.; Shu, L.; Li, X.; Yin, H.; Wang, J.; et al. Host development overwhelms environmental dispersal in governing the ecological succession of zebrafish gut microbiota. NPJ Biofilms Microbiomes 2021, 7, 5. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Jin, X.; Liu, C.; Fan, C.; Guo, L.; Liang, Y.; Zheng, J.; Peng, N. Developmental, Dietary, and Geographical Impacts on Gut Microbiota of Red Swamp Crayfish (Procambarus clarkii). Microorganisms 2020, 8, 1376. [Google Scholar] [CrossRef]
- Wang, S.T.; Meng, X.Z.; Zhang, J.H.; Dai, Y.F.; Shen, Y.; Xu, X.Y.; Wang, R.Q.; Li, J.L. 16S rRNA sequencing analysis of the correlation between the intestinal microbiota and body-mass of grass carp (Ctenopharyngodon idella). Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 35, 100699. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, K.; Wu, J.; Qiu, Q.L.; Yang, K.; Qian, Y.; Zhang, D. Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Appl. Microbiol. Biotechnol. 2015, 99, 6911–6919. [Google Scholar] [CrossRef]
- Duan, Y.; Wang, Y.; Dong, H.; Ding, X.; Liu, Q.; Li, H.; Zhang, J.; Xiong, D. Changes in the Intestine Microbial, Digestive, and Immune-Related Genes of Litopenaeus vannamei in Response to Dietary Probiotic Clostridium butyricum Supplementation. Front. Microbiol. 2018, 9, 2191. [Google Scholar] [CrossRef]
- Fan, J.; Chen, L.; Mai, G.; Zhang, H.; Yang, J.; Deng, D.; Ma, Y. Dynamics of the gut microbiota in developmental stages of Litopenaeus vannamei reveal its association with body weight. Sci. Rep. 2019, 9, 734. [Google Scholar] [CrossRef]
- Yang, H.; Wu, J.; Huang, X.; Zhou, Y.; Zhang, Y.; Liu, M.; Liu, Q.; Ke, S.; He, M.; Fu, H.; et al. ABO genotype alters the gut microbiota by regulating GalNAc levels in pigs. Nature 2022, 606, 358–367. [Google Scholar] [CrossRef]
- Sha, Y.J.; Liu, M.; Wang, B.J.; Jiang, K.Y.; Sun, G.Q.; Wang, L. Gut bacterial diversity of farmed sea cucumbers Apostichopus japonicus with different growth rates. Microbiology 2016, 85, 109–115. [Google Scholar] [CrossRef]
- Alfaro, A.C.; Young, T. Showcasing metabolomic applications in aquaculture: A review. Rev. Aquac. 2016, 10, 135–152. [Google Scholar] [CrossRef]
- Uengwetwanit, T.; Uawisetwathana, U.; Arayamethakorn, S.; Khudet, J.; Chaiyapechara, S.; Karoonuthaisiri, N.; Rungrassamee, W. Multi-omics analysis to examine microbiota, host gene expression and metabolites in the intestine of black tiger shrimp (Penaeus monodon) with different growth performance. PeerJ 2020, 8, e9646. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Huang, L.; Hu, S.; Chen, C.; Huang, X.; Liu, W.; Wang, S.; Zhu, Y.; Zhao, Y.; Zhang, D. Effects of Carbon/Nitrogen Ratio on Growth, Intestinal Microbiota and Metabolome of Shrimp (Litopenaeus vannamei). Front. Microbiol. 2020, 11, 652. [Google Scholar] [CrossRef]
- Chen, B.; Qiu, J.Q.; Wang, Y.X.; Huang, W.; Zhao, H.X.; Zhu, X.F.; Peng, K. Condensed tannins increased intestinal permeability of Chinese seabass (Lateolabrax maculatus) based on microbiome-metabolomics analysis. Aquaculture 2022, 560, 738615. [Google Scholar] [CrossRef]
- Zhang, W.; Pang, A.; Tan, B.; Xin, Y.; Liu, Y.; Xie, R.; Zhang, H.; Yang, Q.; Deng, J.; Chi, S. Tryptophan metabolism and gut flora profile in different soybean protein induced enteritis of pearl gentian groupers. Front. Nutr. 2022, 9, 1014502. [Google Scholar] [CrossRef]
- He, S.; Yu, D.; Li, P.; Zhang, M.; Xing, S.Y.; Sun, C.C.; Li, Z.H. Triphenyltin exposure causes changes in health-associated gut microbiome and metabolites in marine medaka. Environ. Pollut. 2021, 288, 117751. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Shao, J.; Liu, Y.; Wang, H.; Luo, Y.; Chen, L. An Integrated Fecal Microbiome and Metabolomics in T2DM Rats Reveal Antidiabetes Effects from Host-Microbial Metabolic Axis of EtOAc Extract from Sophora flavescens. Oxid. Med. Cell. Longev. 2020, 2020, 1805418. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef]
- Grieneisen, L.; Dasari, M.; Gould, T.J.; Björk, J.R.; Grenier, J.C.; Yotova, V.; Jansen, D.; Gottel, N.; Gordon, J.B.; Learn, N.H.; et al. Gut microbiome heritability is nearly universal but environmentally contingent. Science 2021, 373, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Nelson, C.D.; Driver, J.D.; Elzo, M.A.; Peñagaricano, F.; Jeong, K.C. Host genetics exerts lifelong effects upon hindgut microbiota and its association with bovine growth and immunity. ISME J. 2021, 15, 2306–2321. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.X.; Luo, J.P.; Liu, P.M.; Yi, S.K.; Tang, Q.Y.; Li, J.F.; Xia, Z.L.; Yang, G.L. Characteristics of intestinal microbiota in male morphotypes of the giant freshwater prawn Macrobrachium rosenbergii. Aquaculture 2022, 555, 738200. [Google Scholar] [CrossRef]
- Liu, B.; Liu, B.; Zhou, Q.L.; Sun, C.X.; Song, C.Y.; Zhang, H.M.; Yang, Z.F.; Shan, F. Patterns of bacterial community composition and diversity following the embryonic development stages of Macrobrachium rosenbergii. Aquacult. Rep. 2020, 17, 100372. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Zhang, B.; Lv, Z.; Li, Z.; Wang, W.; Li, G.; Guo, Y. Dietary L-arginine Supplementation Alleviates the Intestinal Injury and Modulates the Gut Microbiota in Broiler Chickens Challenged by Clostridium perfringens. Front. Microbiol. 2018, 9, 1716. [Google Scholar] [CrossRef]
- Fan, J.; Li, X.; Lu, H.; Lin, R.; Aweya, J.J.; Zhang, Y. N-terminal diversity of Litopenaeus vannamei hemocyanin and immunity. Mol. Immunol. 2019, 112, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Meehan, C.J.; Beiko, R.G. A Phylogenomic view of ecological specialization in the Lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol. Evol. 2014, 6, 703–713. [Google Scholar] [CrossRef]
- Silva, B.C.D.; Vieira, F.D.N.; Mouriño, J.L.P.; Bolivar, N.; Seiffert, W.Q. Butyrate and propionate improve the growth performance of Litopenaeus vannamei. Aquac. Res. 2016, 47, 612–623. [Google Scholar] [CrossRef]
- Li, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and Comparison of Microbiota in the Gastrointestinal Tracts of the Goat (Capra hircus) During Preweaning Development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef]
- Catalán, N.; Villasante, A.; Wacyk, J.; Ramírez, C.; Romero, J. Fermented Soybean Meal Increases Lactic Acid Bacteria in Gut Microbiota of Atlantic Salmon (Salmon salar). Probiotics Antimicrob. Proteins 2018, 10, 566–576. [Google Scholar] [CrossRef]
- Villasante, A.; Catalán, N.; Rojas, R.; Lohrmann, K.B.; Romero, J. Microbiota of the Digestive Gland of Red Abalone (Haliotis rufescens) Is Affected by Withering Syndrome. Microorganisms 2020, 8, 1411. [Google Scholar] [CrossRef] [PubMed]
- El-Sohemy, A. Nutrigenetics. Nutr.-Oppor. Asia 2007, 60, 25–30. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, X.W.; Liang, G.; Pan, C.W. Metabolomics in Glaucoma: A Systematic Review. Investig. Ophthalmol. Vis. Sci. 2021, 62, 9. [Google Scholar] [CrossRef]
- Han, T.; Cai, D.; Geng, S.; Wang, Y.; Zhen, H.; Wu, P. Effect of whey protein on plasma amino acids in diabetic mice. Exp. Ther. Med. 2013, 6, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, H.; Liu, Q.; Gao, Y.; Dai, J.; Bao, B.; Zhang, L.; Ding, A. Cellular Metabolomics Revealed the Cytoprotection of Amentoflavone, a Natural Compound, in Lipopolysaccharide-Induced Injury of Human Umbilical Vein Endothelial Cells. Int. J. Mol. Sci. 2016, 17, 1514. [Google Scholar] [CrossRef]
- Sarafidis, K.; Chatziioannou, A.C.; Thomaidou, A.; Gika, H.; Mikros, E.; Benaki, D.; Diamanti, E.; Agakidis, C.; Raikos, N.; Drossou, V.; et al. Urine metabolomics in neonates with late-onset sepsis in a case-control study. Sci. Rep. 2017, 7, 45506. [Google Scholar] [CrossRef]
- Muñoz-Esparza, N.C.; Latorre-Moratalla, M.L.; Comas-Basté, O.; Toro-Funes, N.; Veciana-Nogués, M.T.; Vidal-Carou, M.C. Polyamines in Food. Front. Nutr. 2019, 6, 108. [Google Scholar] [CrossRef]
- Niechcial, A.; Butter, M.; Manz, S.; Obialo, N.; Bäbler, K.; Lely, L.V.D.; Lang, S.; Gottier, C.; McCole, D.F.; Scharl, M.; et al. Presence of PTPN2 SNP rs1893217 Enhances the Anti-inflammatory Effect of Spermidine. Inflamm. Bowel Dis. 2020, 26, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wu, X.; Jia, G.; Chen, X.; Zhao, H.; Wang, J.; Wu, C.; Cai, J. Arginine: New Insights into Growth Performance and Urinary Metabolomic Profiles of Rats. Molecules 2016, 21, 1142. [Google Scholar] [CrossRef]
- Xiao, Y.P.; Wu, T.X.; Hong, Q.H.; Sun, J.M.; Chen, A.G.; Yang, C.M.; Li, X.Y. Response to weaning and dietary L-glutamine supplementation: Metabolomic analysis in piglets by gas chromatography/mass spectrometry. J. Zhejiang Univ. Sci. B. 2012, 13, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Tsonaka, R.; Signorelli, M.; Sabir, E.; Seyer, A.; Hettne, K.; Aartsma-Rus, A.; Spitali, P. Longitudinal metabolomic analysis of plasma enables modeling disease progression in Duchenne muscular dystrophy mouse models. Hum. Mol. Genet. 2020, 29, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Boison, D. Adenosine kinase: Exploitation for therapeutic gain. Pharmacol. Rev. 2013, 65, 906–943. [Google Scholar] [CrossRef]
- Arin, R.M.; Vallejo, A.I.; Rueda, Y.; Fresnedo, O.; Ochoa, B. Expression of Adenosine A2B Receptor and Adenosine Deaminase in Rabbit Gastric Mucosa ECL Cells. Molecules 2017, 22, 625. [Google Scholar] [CrossRef]
- Watson, D.G.; Pomeroy, P.P.; Al-Tannak, N.F.; Kennedy, M.W. Stockpiling by pups and self-sacrifice by their fasting mothers observed in birth to weaning serum metabolomes of Atlantic grey seals. Sci. Rep. 2020, 10, 7465. [Google Scholar] [CrossRef]
- Yang, H.; Xu, D.; Zhuo, Z.; Hu, J.; Lu, B. Transcriptome and gene expression analysis of Rhynchophorus ferrugineus (Coleoptera: Curculionidae) during developmental stages. PeerJ 2020, 8, e10223. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qiao, L.; An, L.; Wang, W.; Liu, J.; Ren, Y.; Pan, Y.; Jing, J.; Liu, W. Transcriptome analysis of adipose tissues from two fat-tailed sheep breeds reveals key genes involved in fat deposition. BMC Genom. 2018, 19, 338. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Liu, Y.; Zhang, W.; Zou, W. Role of diet on intestinal metabolites and appetite control factors in SD rats. Exp. Ther. Med. 2020, 20, 2665–2674. [Google Scholar] [CrossRef]
- Sheraj, I.; Guray, N.T.; Banerjee, S. A pan-cancer transcriptomic study showing tumor specific alterations in central metabolism. Sci. Rep. 2021, 11, 13637. [Google Scholar] [CrossRef]
- Muroya, S.; Oe, M.; Ojima, K.; Watanabe, A. Metabolomic approach to key metabolites characterizing postmortem aged loin muscle of Japanese Black (Wagyu) cattle. Asian-Australas. J. Anim. Sci. 2019, 32, 1172–1185. [Google Scholar] [CrossRef]
- Abela, L.; Spiegel, R.; Crowther, L.M.; Klein, A.; Steindl, K.; Papuc, S.M.; Joset, P.; Zehavi, Y.; Rauch, A.; Plecko, B.; et al. Plasma metabolomics reveals a diagnostic metabolic fingerprint for mitochondrial aconitase (ACO2) deficiency. PLoS ONE 2017, 12, e0176363. [Google Scholar] [CrossRef]
- Nishii, M.; Ito, S.; Katayama, N.; Osanai, T. Biochemical elucidation of citrate accumulation in Synechocystis sp. PCC 6803 via kinetic analysis of aconitase. Sci. Rep. 2021, 11, 17131. [Google Scholar] [CrossRef]
- Hien, T.T.T.; Be, T.T.; Lee, C.M.; Bengtson, D.A. Development of formulated diets for snakehead (Channa striata and Channa micropeltes): Can phytase and taurine supplementation increase use of soybean meal to replace fish meal? Aquaculture 2015, 448, 334–340. [Google Scholar] [CrossRef]
- Al-Feky, S.S.A.; El-Sayed, A.F.M.; Ezzat, A.A. Dietary taurine enhances growth and feed utilization in larval Nile tilapia (Oreochromis niloticus) fed soybean meal-based diets. Aquac. Nutr. 2016, 22, 457–464. [Google Scholar] [CrossRef]
- Cheng, C.H.; Guo, Z.X.; Wang, A.L. The protective effects of taurine on oxidative stress, cytoplasmic free-Ca2+ and apoptosis of pufferfish (Takifugu obscurus) under low temperature stress. Fish Shellfish. Immunol. 2018, 77, 457–464. [Google Scholar] [CrossRef]
- Hawkyard, M.; Laurel, B.; Barr, Y.; Hamre, K.; Langdon, C. Evaluation of liposomes for the enrichment of rotifers (Brachionus sp.) with taurine and their subsequent effects on the growth and development of northern rock sole (Lepidopsetta polyxystra) larvae. Aquaculture 2015, 441, 118–125. [Google Scholar] [CrossRef]
- Flint, H.J.; Bayer, E.A.; Rincon, M.T.; Lamed, R.; White, B.A. Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nat. Rev. Microbiol. 2008, 6, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Seenivasan, C.; Bhavan, P.S.; Radhakrishnan, S. Effect of probiotics (BinifitTM) on survival, growth, biochemical constituents and energy budget of the freshwater prawn Macrobrachium rosenbergii post larvae. Aquaculture 2011, 41, 5919–5927. [Google Scholar]
- Liu, X.; Mao, B.; Gu, J.; Wu, J.; Cui, S.; Wang, G.; Zhao, J.; Zhang, H.; Chen, W. Blautia—A new functional genus with potential probiotic properties? Gut Microbes 2021, 13, 1875796. [Google Scholar] [CrossRef]
- Hosomi, K.; Saito, M.; Park, J.; Murakami, H.; Shibata, N.; Ando, M.; Nagatake, T.; Konishi, K.; Ohno, H.; Tanisawa, K.; et al. Oral administration of Blautia wexlerae ameliorates obesity and type 2 diabetes via metabolic remodeling of the gut microbiota. Nat. Commun. 2022, 13, 4477. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | FBW (g) | FBL (cm) | WG (%) | SGR (%/d) |
|---|---|---|---|---|
| Group H | 41.03 ± 1.44 A | 11.28 ± 0.19 A | 1616.82 ± 60.08 A | 3.09 ± 0.04 A |
| Group M | 37.46 ± 0.73 B | 10.99 ± 0.11 B | 1467.45 ± 30.54 B | 2.99 ± 0.02 B |
| Group L | 33.34 ± 1.97 C | 10.59 ± 0.24 C | 1294.82 ± 82.31 C | 2.86 ± 0.07 C |
| Phylum | Family | Genus | Species | Group H | Group M | Group L | |||
|---|---|---|---|---|---|---|---|---|---|
| Mean (%) | S.D. (%) | Mean (%) | S.D. (%) | Mean (%) | S.D. (%) | ||||
| Proteobacteria | 83.66 | 8.242 | 88.29 | 6.424 | 92.80 | 2.199 | |||
| Firmicutes | 14.20 | 8.656 | 9.665 | 5.391 | 4.370 | 2.558 | |||
| Bacteroidota | 0.397 | 0.335 | 0.408 | 0.506 | 0.797 | 0.933 | |||
| Desulfobacterota | 0.204 | 0.221 | 0.012 | 0.009 | 0.006 | 0.005 | |||
| Firmicutes | Lactobacillaceae | 0.470 | 0.705 | 0.021 | 0.010 | 0.006 | 0.005 | ||
| Firmicutes | Lachnospiraceae | 0.502 | 0.333 | 0.027 | 0.025 | 0.007 | 0.007 | ||
| Firmicutes | Peptostreptococcaceae | 0.136 | 0.111 | 0.036 | 0.036 | 0.010 | 0.004 | ||
| Firmicutes | Lactobacillaceae | Lactobacillus | 0.431 | 0.700 | 0.007 | 0.005 | 0.001 | 0.001 | |
| Firmicutes | Lachnospiraceae | CHKCI001 | 0.188 | 0.221 | —— | —— | —— | —— | |
| Firmicutes | Peptostreptococcaceae | Romboutsia | 0.111 | 0.114 | 0.003 | 0.003 | 0.001 | 0.001 | |
| Firmicutes | Lachnospiraceae | [Ruminococcus]_torques_group | 0.095 | 0.102 | 0.0002 | 0.001 | —— | —— | |
| Firmicutes | Lachnospiraceae | Blautia | 0.043 | 0.022 | 0.003 | 0.005 | 0.0005 | 0.001 | |
| Firmicutes | Ruminococcaceae | Faecalibacterium | 0.057 | 0.057 | 0.007 | 0.007 | 0.005 | 0.005 | |
| Firmicutes | Butyricicoccaceae | Butyricicoccus | 0.014 | 0.016 | —— | —— | —— | —— | |
| Firmicutes | Streptococcaceae | Streptococcus | 0.017 | 0.010 | 0.010 | 0.008 | 0.004 | 0.004 | |
| Proteobacteria | Enterobacteriaceae | Enterobacter | 2.045 | 2.123 | 1.426 | 0.327 | 1.356 | 0.554 | |
| Proteobacteria | Enterobacteriaceae | Escherichia-Shigella | 0.156 | 0.291 | 0.009 | 0.009 | 0.003 | 0.002 | |
| Proteobacteria | Vibrionaceae | Vibrio | 0.018 | 0.017 | 0.014 | 0.010 | 0.005 | 0.004 | |
| Bacteroidota | Rikenellaceae | Rikenellaceae_RC9_gut_group | 0.129 | 0.246 | —— | —— | —— | —— | |
| Bacteroidota | Bacteroidaceae | Bacteroides | 0.037 | 0.041 | 0.002 | 0.002 | 0.001 | 0.001 | |
| Desulfobacterota | Desulfovibrionaceae | Desulfovibrio | 0.126 | 0.159 | 0.002 | 0.002 | 0.001 | 0.002 | |
| Firmicutes | Peptostreptococcaceae | Romboutsia | Romboutsia_ilealis | 0.111 | 0.114 | 0.003 | 0.003 | 0.001 | 0.001 |
| Mode | Pairwise Comparison | Total Differential Metabolites Number | Up/Downregulated Differential Metabolites Number | Identification Level 1 Differential Metabolites Number | Identification Level 2 Differential Metabolites Number | ||
|---|---|---|---|---|---|---|---|
| Upregulated | Downregulated | Upregulated | Downregulated | ||||
| ESI+ | Group L vs. Group H | 338 | 169/169 | 3 | 8 | 9 | 10 |
| Group L vs. group M | 402 | 256/146 | 6 | 8 | 23 | 13 | |
| Group M vs. Group H | 433 | 191/242 | 5 | 8 | 9 | 22 | |
| ESI− | Group L vs. Group H | 107 | 60/47 | 1 | 0 | 4 | 5 |
| Group L vs. group M | 168 | 87/81 | 2 | 0 | 5 | 0 | |
| Group M vs. Group H | 153 | 94/59 | 9 | 1 | 13 | 6 | |
| Name | Pathway | Group H | Group M | Group L |
|---|---|---|---|---|
| Mean | Mean | Mean | ||
| Spermidine | Arginine and proline metabolism | 7,568,085.52 | 503,668.63 | 297,921.56 |
| L-citrulline | Arginine biosynthesis | 12,351,334.96 | 28,501,478.19 | 50,349,665.38 |
| 4-oxoproline | Arginine and proline metabolism | 34,504,224.16 | 21,513,189.36 | 14,195,014.97 |
| Adenosine | Purine metabolism | 20,948,251.26 | 16,955,727.72 | 5,034,361.71 |
| N-acetyl-l-phenylalanine | Phenylalanine metabolism | 16,939,441.79 | 9,720,963.52 | 8,590,198.80 |
| Tryptophol | Tryptophan metabolism | 3,882,503.94 | 2,171,436.62 | 1,993,910.23 |
| 8(s)-hydroxy-(5z,9e,11z,14z)-eicosatetraenoic acid | Arachidonic acid metabolism | 12,940,493.38 | 12,410,918.29 | 8,284,469.84 |
| 13(s)-hotre | alpha-Linolenic acid metabolism | 9,965,759.94 | 5,687,309.98 | 5,470,815.92 |
| Hydroxylysine | Lysine degradation | 1,790,922.13 | 1,040,770.98 | 576,601.17 |
| P-dmea | Glycerophospholipid metabolism | 2,420,117.054 | 5,503,074.12 | 6,928,635.32 |
| Maleamic acid | Nicotinate and nicotinamide metabolism | 3,819,384.43 | 7,534,743.65 | 9,223,492.68 |
| Trimethylamine n-oxide | Metabolic pathways | 11,407,950.24 | 9,534,357.45 | 2,708,587.02 |
| Cytosine | Pyrimidine metabolism | 26,885,730.18 | 32,999,513.02 | 67,604,815.23 |
| Creatinine | Arginine and proline metabolism | 24,423,233.36 | 18,256,455.97 | 15,024,899.59 |
| 4-guanidinobutyric acid | Arginine and proline metabolism | 4,072,488.34 | 4,784,183.04 | 7,288,469.76 |
| 5-guanidino-2-oxopentanoic acid | Arginine and proline metabolism | 5,655,838.31 | 8,563,793.46 | 10,158,770.44 |
| N-[(5s)-5-amino-5-carboxypentanoyl] cysteinyl-d-valine | Metabolic pathways | 2,077,361.39 | 2,046,182.39 | 1,352,700.594 |
| 3-methoxytyramine | Tyrosine metabolism | 622,589.31 | 1,369,607.54 | 1,611,085.97 |
| (9cis)-retinal | Retinol metabolism | 13,138,741.23 | 20,809,948.49 | 30,180,669.50 |
| 4-hydroxy-3-octaprenylbenzoic acid | Ubiquinone and another terpenoid-quinone biosynthesis | 7,617,111.30 | 20,111,478.28 | 20,269,682.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, X.; Peng, X.; Du, T.; Xia, Z.; Gao, Q.; Tang, Q.; Yi, S.; Yang, G. Alterations of the Gut Microbiota and Metabolomics Associated with the Different Growth Performances of Macrobrachium rosenbergii Families. Animals 2023, 13, 1539. https://doi.org/10.3390/ani13091539
Lan X, Peng X, Du T, Xia Z, Gao Q, Tang Q, Yi S, Yang G. Alterations of the Gut Microbiota and Metabolomics Associated with the Different Growth Performances of Macrobrachium rosenbergii Families. Animals. 2023; 13(9):1539. https://doi.org/10.3390/ani13091539
Chicago/Turabian StyleLan, Xuan, Xin Peng, Tingting Du, Zhenglong Xia, Quanxin Gao, Qiongying Tang, Shaokui Yi, and Guoliang Yang. 2023. "Alterations of the Gut Microbiota and Metabolomics Associated with the Different Growth Performances of Macrobrachium rosenbergii Families" Animals 13, no. 9: 1539. https://doi.org/10.3390/ani13091539