
Full-Length Transcriptome and Gene Expression Analysis of Different Ovis aries Adipose Tissues Reveals Transcript Variants Involved in Lipid Biosynthesis
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sequencing Library Preparation
2.3. Initial Analysis of Full-Length Sequencing Data
2.4. Identification and Annotation of Novel Genes and Isoforms
2.5. Recognition of Alternative Splicing, Fusion Genes, and Alternative Polyadenylation
2.6. Analysis of Short-Length Sequencing Data
2.7. Validation of Sequencing Accuracy
3. Results
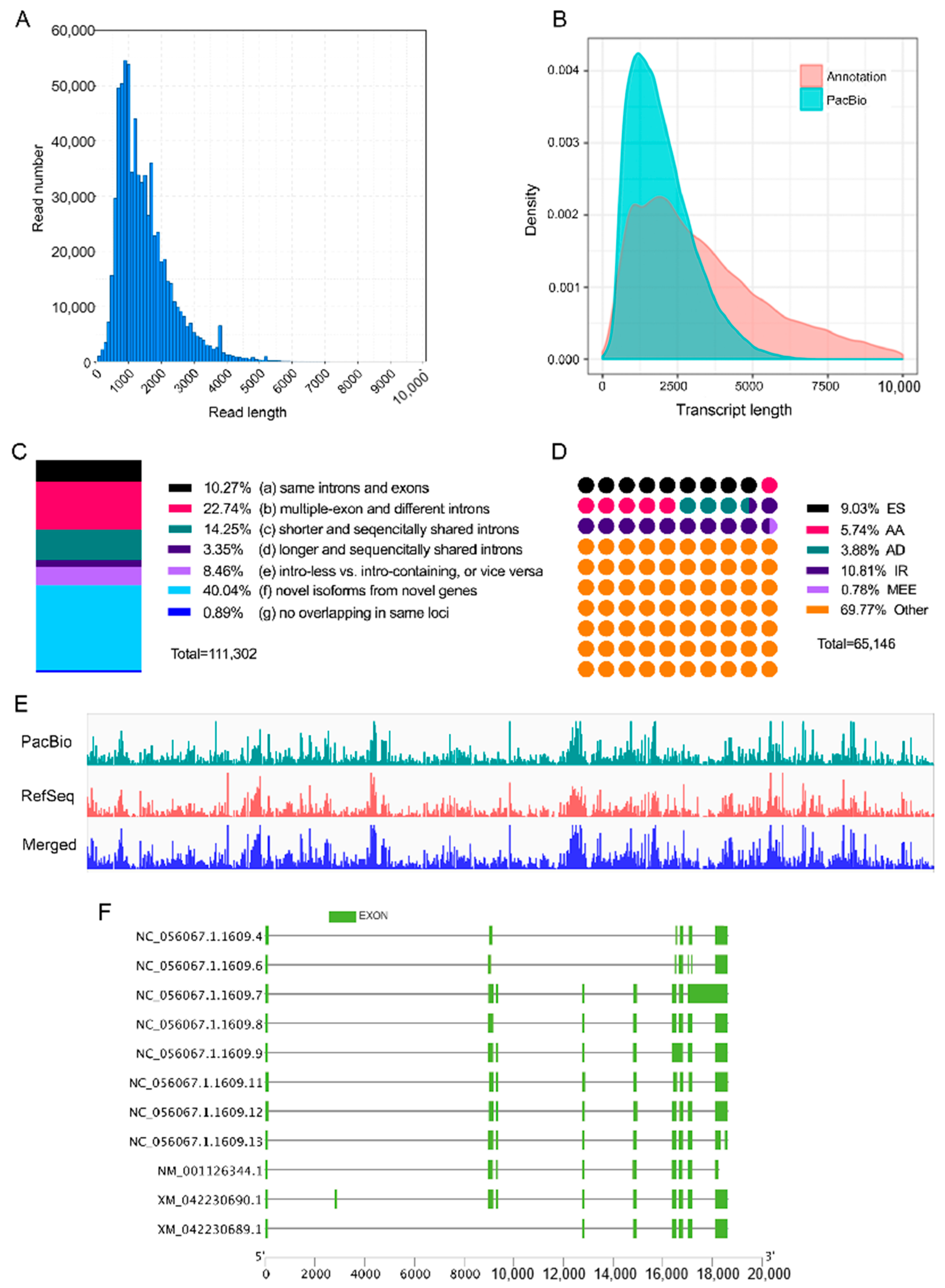
3.1. Overall Description of Full-Length Sequencing and Error Correction
3.2. Loci and Isoform Identification in Full-Length Sequencing
3.3. Transcriptome Diversity
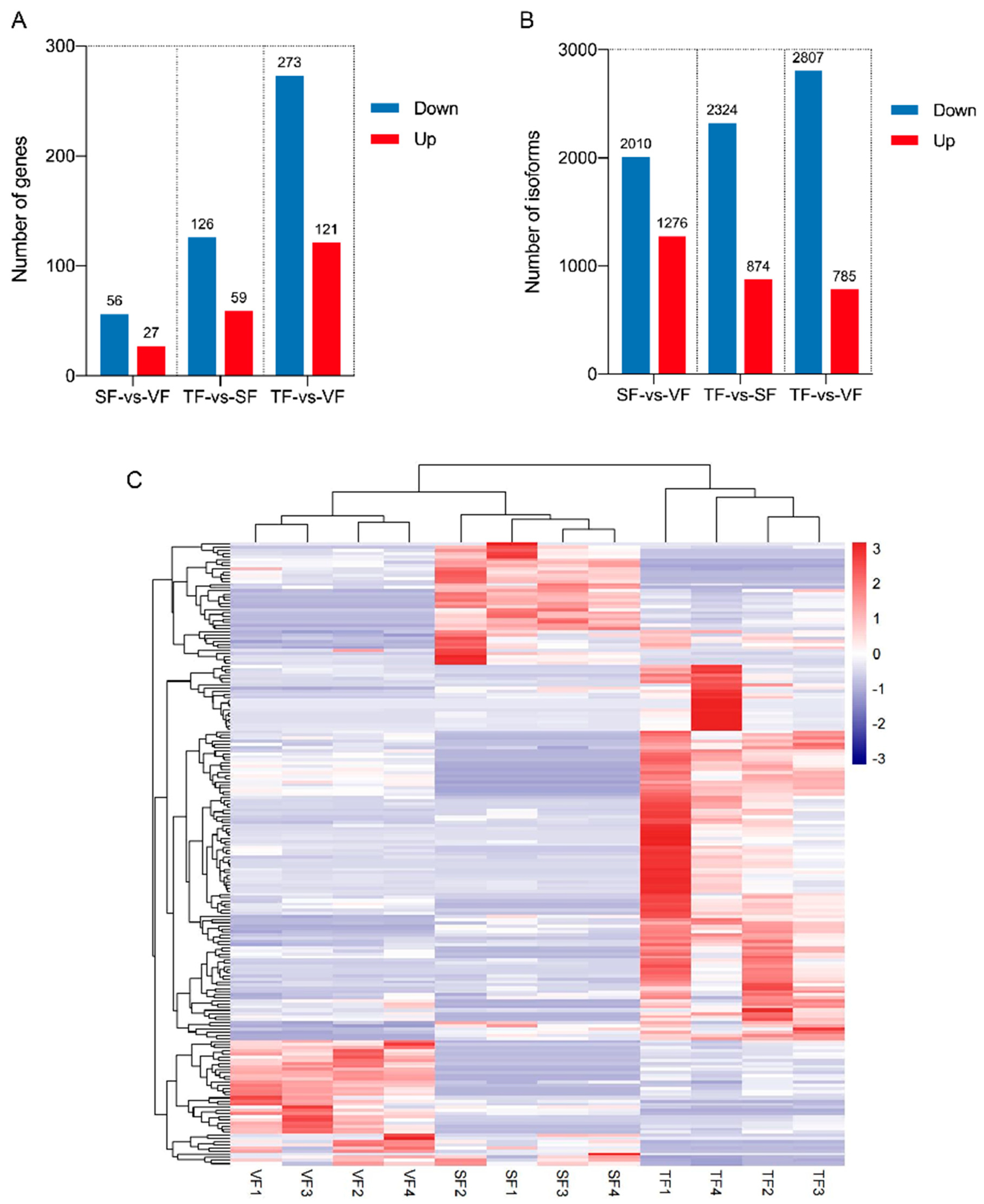
3.4. Overall Description of Short-Read Sequencing Data
3.5. DEGs and DEIs
3.6. Functional Annotation of DEGs and DEIs
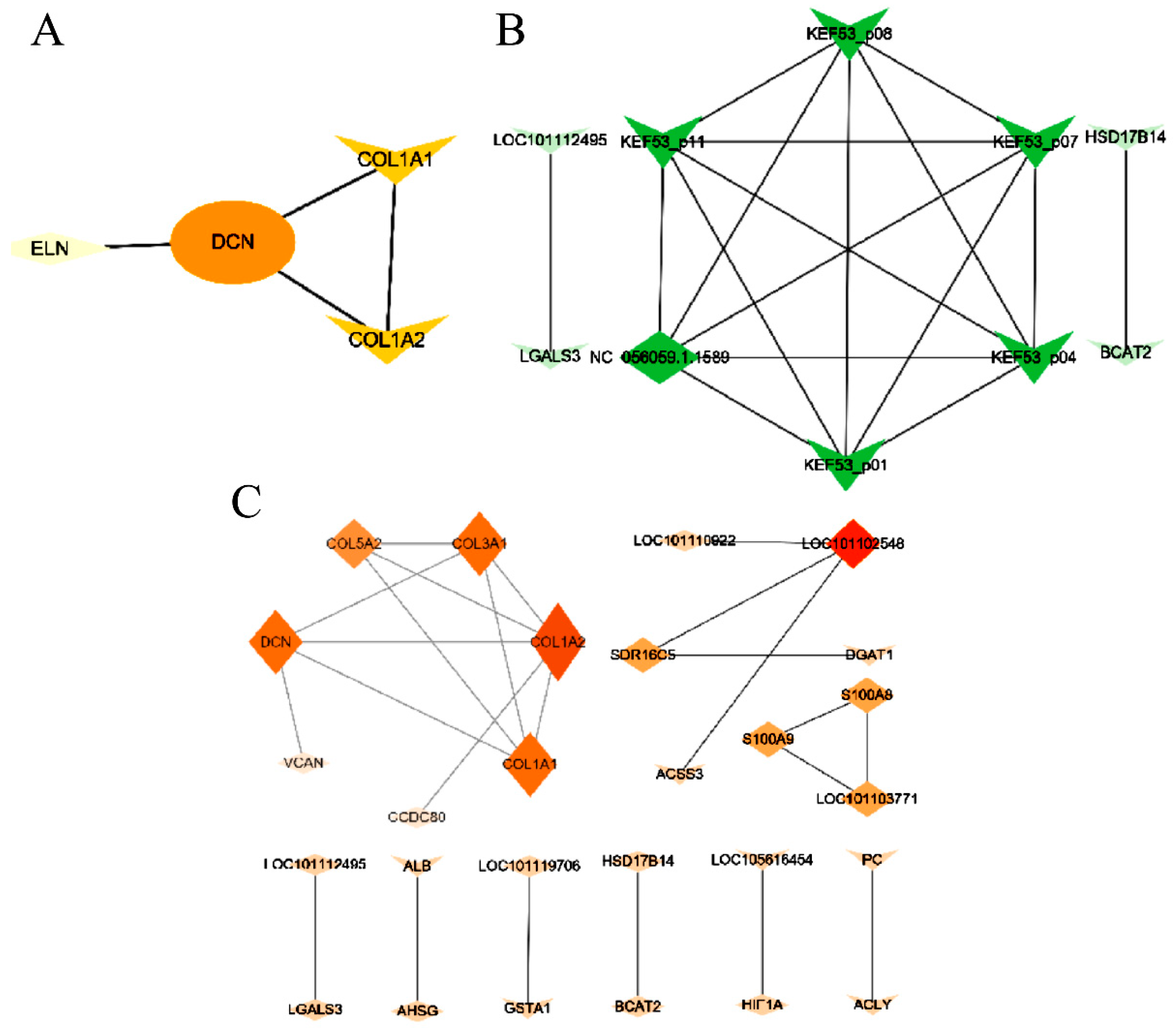
3.7. Interactions between DEGs
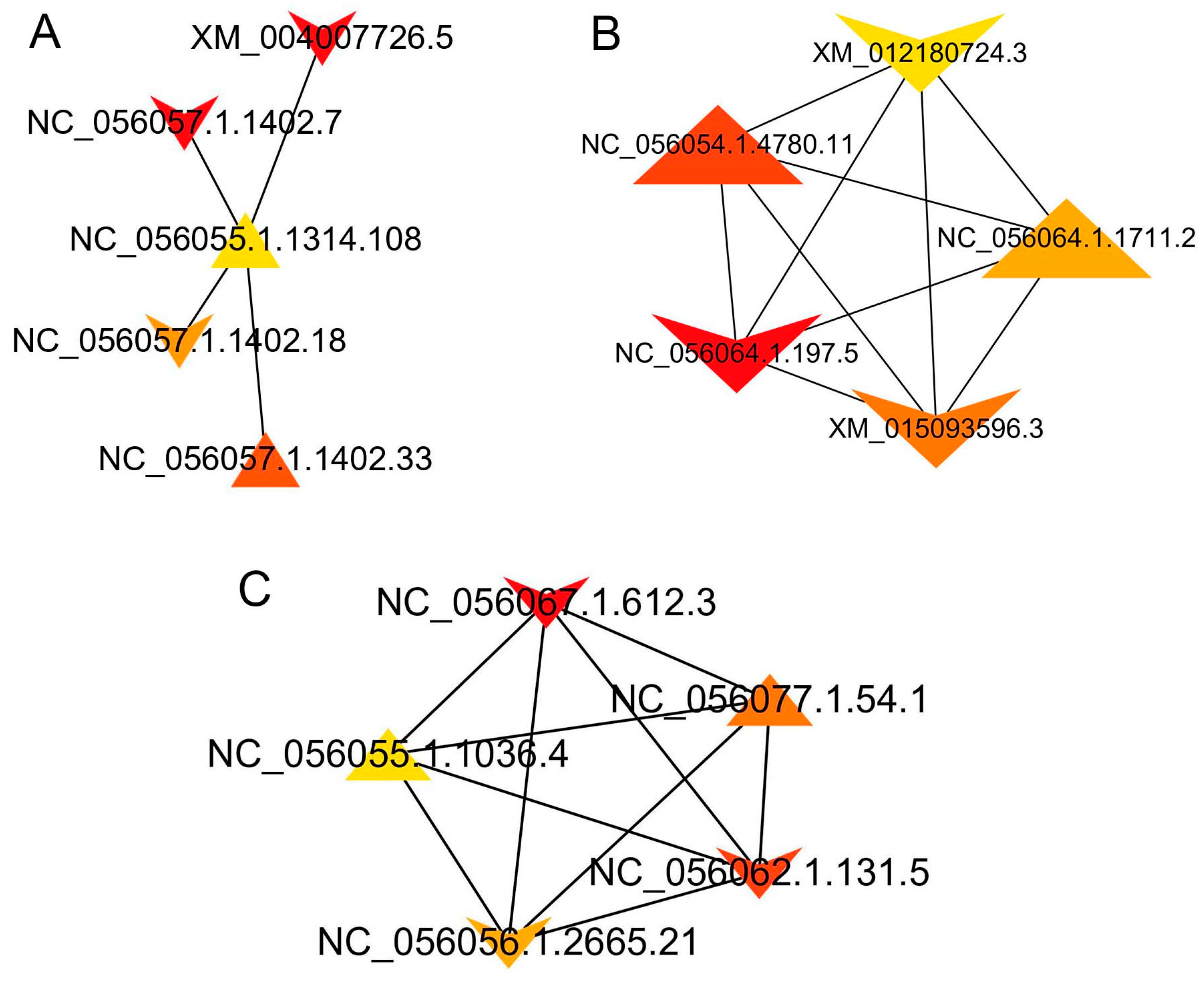
3.8. Interactions between DEIs
3.9. qRT-PCR Validation of DEGs
4. Discussion
4.1. Transcriptome Sequencing Data Quality
4.2. Enrichment and Roles of DEGs in Fat Metabolism
4.3. Enrichment and Roles of DEIs in Fat Metabolism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chikwanha, O.C.; Vahmani, P.; Muchenje, V.; Dugan, M.E.R.; Mapiye, C. Nutritional Enhancement of Sheep Meat Fatty Acid Profile for Human Health and Wellbeing. Food Res. Int. 2018, 104, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Yang, X.; Lian, Z.; Liu, G.; Sun, B.; Guo, Y.; Li, Y.; Liu, D. A Comparative Study on the Improvements by Crossbreeding of Dorper Sheep and Australian White Sheep with Hu Sheep. Anim. Husb. Vet. Med. 2018, 50, 6–10. [Google Scholar]
- Yuan, Z.; Ge, L.; Sun, J.; Zhang, W.; Wang, S.; Gao, X.; Sun, W. Integrative Aanalysis of Iso-Seq and RNA-seq Data Reveals Transcriptome Complexity and Differentially Expressed Transcripts in Sheep Tail Fat. PeerJ 2021, 9, e12454. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Y.; Hussain, T.; Dai, C.; Li, J.; Huang, P.; Li, Y.; Ding, X.; Huang, J.; Ji, F.; et al. Effects of Dietary Energy Level on Growth Performance, Blood Parameters and Meat Quality in Fattening Male Hu Lambs. J. Anim. Physiol. Anim. Nutr. 2020, 104, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Mao, Y.; Duan, D.; Dan, J.; Cheng, X.; Guo, H.; Wang, H.; Meng, D. Progressive Crossing Experiment of Duper Sheep and Hu Sheep for Meat. Heilongjiang Anim. Husbandry Vet. Sci. 2018, 12, 74–76. [Google Scholar]
- Wang, J.; Song, Y.; Cheng, X.; Yu, F.; Cao, B. Preliminary Study on the Germplasm Characteristics of Duper Sheep. Chin. J. Anim. Sci. 2005, 11, 323–325. [Google Scholar]
- Gorni, C.; Garion, C.; Iacuaniello, S.; Castiglioni, B.; Stella, A.; Restelli, G.L.; Pagnacco, G.; Mariani, P. Transcriptome Analysis to Identify Differential Gene Expression Affecting Meat Quality in Heavy Italian Pigs. Anim. Genet. 2010, 42, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Srikanth, K.; Lim, D.; Park, M.; Hur, T.; Kemp, S.; Dessie, T.; Kim, M.S.; Lee, S.; Pas, M.F.W.T.; et al. Comparative Transcriptome Analysis of Ethiopian Indigenous Chickens from Low and High Altitudes under Heat Stress Condition Reveals Differential Immune Response. Anim. Genet. 2018, 50, 42–53. [Google Scholar] [CrossRef]
- Du, L.; Chang, T.; An, B.; Liang, M.; Deng, T.; Li, K.; Cao, S.; Du, Y.; Gao, X.; Xu, L.; et al. Transcriptomics and Lipid Metabolomics Analysis of Subcutaneous, Visceral and Abdominal Adipose Tissues of Beef Cattle. Genes 2022, 14, 37. [Google Scholar] [CrossRef]
- Pan, Y.; Chen, S.; Niu, S.; Bi, X.; Qiao, L.; Yang, K.; Liu, J.; Liu, W. Hybrid Sequencing in Different Types of Goat Skeletal Muscles Reveals Genes Regulating Muscle Development and Meat Quality. Animals 2021, 11, 2906. [Google Scholar] [CrossRef]
- Ahmad, S.; Drag, M.H.; Salleh, S.M.; Cai, Z.; Nielsen, M.O. Transcriptomics Analysis of Differentially Expressed Genes in Subcutaneous and Perirenal Adipose Tissue of Sheep as Affected by Their Pre- and Early Postnatal Malnutrition Histories. BMC Genom. 2021, 22, 338. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Jing, J.; Qiao, L.; Liu, J.; An, L.; Li, B.; Ren, D.; Liu, W. MiRNA-Seq Reveals That miR-124-3p Inhibits Adipogenic Differentiation of the Stromal Vascular Fraction in Sheep via Targeting C/EBPα. Domest. Anim. Endocrinol. 2018, 65, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Fei, X.; Li, T.; Lu, Z.; Chu, M.; Di, R.; He, X.; Wang, X.; Wei, C. Transcriptome Study Digs out BMP2 Involved in Adipogenesis in Sheep Tails. BMC Genom. 2022, 23, 457. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xu, D.; Zhuo, Z.; Hu, J.; Lu, B. SMRT sequencing of the full-length transcriptome of the Rhynchophorus ferrugineus (Coleoptera: Curculionida). PeerJ 2020, 8, e91333. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.M.; Sanjana, H.; Nguyen, L.T.; Cheng, Y.; Moore, S.S.; Hayes, B.J. Extensive Variation in Gene Expression is Revealed in 13 Fertility-Related Genes Using RNA-Seq, ISO-Seq, and CAGE-Seq From Brahman Cattle. Front Genet 2022, 13, 784663. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, C.M.; Noble, J.D.; Pietras, A.; Moler, P.; Austin, J.D. Iso-seq Analysis and Functional Anotation of The Santa Fe Cave Crayfish (Procambarus erythrops) Transcriptome. Mar. Genom. 2021, 5, 100842. [Google Scholar] [CrossRef] [PubMed]
- Shields, E.J.; Sorida, M.; Sheng, L.; Sieriebriennikov, B.; Ding, L.; Bonasio, R. Genome Annotation with Long RNA Reads Reveals New Patterns of Gene Expression and Improves Single-cell Analyses in An Ant Brain. BMC Biol. 2021, 19, 254. [Google Scholar] [CrossRef]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and Efficient Long Read Error Correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A Genomic Mapping and Alignment Program for mRNA and EST Sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Kryolov, D.M.; Mazmuder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG Database: An Updated Version Includes Eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Jung, E.; Bairoch, A. SWISS-PROT: Connecting biomolecular knowledge via a protein database. Curr. Issues Mol. Biol. 2001, 3, 47–55. [Google Scholar] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool Using an Alignment-Free Logistic Regression Model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of Exon Skipping Events Differentiate Among Splicing Patterns in Sixteen Human Tissues. F1000Res 2013, 2, 188. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A Survey of the Sorghum Transcriptome Using Single-Molecule Long Reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification From RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zhang, H.M.; Liu, T.; Liu, C.J.; Song, S.; Zhang, X.; Liu, W.; Jia, H.; Xue, Y.; Guo, A.Y. AnimalTFDB 2.0: A Resource for Expression, Prediction and Functional Study of Animal Transcription Factors. Nucleic Acids Res. 2015, 43, D76–D81. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING Database in 2017: Quality-Controlled Protein–Protein Association Networks, Made Broadly Accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Zhou, G.; Zhou, S.; Zeng, J.; Wang, X.; Jiang, Y.; Yang, Y.; Chen, Y. Comparative transcriptome analysis reveals potentially novel roles of Homeobox genes in adipose depositon in fat-tailed sheep. Sci. Rep. 2017, 7, 14491. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Qin, M.; Zhang, Y.; Xue, H.; Li, S.; Chen, W.; Zeng, Y. Identification and functional prediction of long non-coding RNAs related to skeletal muscle development in Duroc pigs. Anim. Biosci. 2022, 35, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Qiao, M.; Guo, M.; Wu, H.; Wu, J.; Wang, D.; Liu, G.; Mei, S.; Pemg, X. Single Nucleotide Polymorphism of Porcine ACADL Gene and Its Association with Fat Deposition Trait. Hubei Agric. Sci. 2018, 57, 161–163. [Google Scholar] [CrossRef]
- Corominas, J.; Ramayo-Caldas, Y.; Castelló, A.; Muñoz, M.; Ibáñez-Escriche, N.; Folch, J.M.; Ballester, M. Evaluation of the Porcine ACSL4 Gene as a Candidate Gene for Meat Quality Traits in Pigs. Anim. Genet. 2012, 43, 714–720. [Google Scholar] [CrossRef]
- Jiang, Z.; Michal, J.J.; Tobey, D.J.; Daniels, T.F.; Rule, D.C.; Macneil, M.D. Significant Associations of Stearoyl-CoA Desaturase (SCD1) Gene with Fat Deposition and Composition in Skeletal Muscle. Int. J. Biol. Sci. 2008, 4, 345–351. [Google Scholar] [CrossRef]
- Adapala, V.J.; Adedokun, S.A.; Considine, R.V.; Ajuwon, K.M. Acute Inflammation Plays a Limited Role in the Regulation of Adipose Tissue COL1A1 Protein Abundance. J. Nutr. Biochem. 2012, 23, 567–572. [Google Scholar] [CrossRef]
- Mansoor, M.A.; Seljeflot, I.; Arnesen, H.; Knudsen, A.; Bates, C.J.; Mishra, G.; Larsen, T.W. Endothelial Cell Adhesion Molecules in Healthy Adults during Acute Hyperhomocysteinemia and Mild Hypertriglyceridemia. Clin. Biochem. 2004, 37, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, R.; Kishida, K.; Nakatsuji, H.; Nakagawa, T.; Funahashi, T.; Shimomura, I. High circulating levels of S100A8/A9 complex (calprotectin) in male Japanese with abdominal adiposity and dysregulated expression of S100A8 and S100A9 in adipose tissues of obese mice. Biochem. Biophys. Res. Commun. 2012, 419, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Walenta, E. Omega-3 Fatty Acids and FFAR4. Front. Endocrinol. 2014, 5, 115. [Google Scholar] [CrossRef] [PubMed]
- Husted, A.S.; Ekberg, J.H.; Tripp, E.; Nissen, T.A.D.; Meijnikman, S.; O’Brien, S.L.; Ulven, T.; Acherman, Y.; Bruin, S.C.; Nieuwdorp, M.; et al. Autocrine Negative Feedback Regulation of Lipolysis Through Sensing of NEFAs by FFAR4/GPR120 in WAT. Mol. Metab. 2020, 42, 101103. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Zhang, Y.; Baye, T.M.; Gawrieh, S.; Cole, R.; Blangero, J.; Carless, M.A.; Curran, J.E.; Dyer, T.D.; Abraham, L.J.; et al. INSIG1 Influences Obesity-related Hypertriglyceridemia in Humans. J. Lipid Res. 2010, 51, 701–708. [Google Scholar] [CrossRef]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerbäck, S.; et al. Functional Brown Adipose Tissue in Healthy Adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef]
- Lim, S.; Honek, J.; Xue, Y.; Seki, T.; Cao, Z.; Andersson, P.; Yang, X.; Hosaka, K.; Cao, Y. Cold-Induced Activation of Brown Adipose Tissue and Adipose Angiogenesis in Mice. Nat. Protoc. 2012, 7, 606–615. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, J.; Dai, H.; Duan, Y.; An, Y.; Shi, L.; Lv, Y.; Li, H.; Wang, C.; Ma, Q.; et al. Brown and Beige Adipose Tissue: A Novel Therapeutic Strategy for Obesity and Type 2 Diabetes Mellitus. Adipocyte 2021, 10, 48–65. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Liu, P.; Yang, F.; Wang, X.; Zheng, W.; Sun, W. Hesperetin Ameliorates Hepatic Oxidative Stress and Inflammation Via the PI3K/AKT-Nrf2-ARE Pathway in Oleic Acid-induced HepG2 Cells and A Rat Model of High-fat Diet-induced NAFLD. Food Funct. 2021, 12, 3898–3918. [Google Scholar] [CrossRef]
- Yang, H.; Cao, Q.; Xiong, X.; Zhao, P.; Shen, D.; Zhang, Y.; Zhang, N. Fluoxetine Regulates Glucose and Lipid Metabolism Via the PI3K-AKT Signaling Pathway in Diabetic Rats. Mol. Med. Rep. 2020, 22, 3073–3080. [Google Scholar] [CrossRef]
- Liu, D.D.; Han, C.C.; Wan, H.F.; He, F.; Xu, H.Y.; Wei, S.H.; Du, X.H.; Xu, F. Effects of Inhibiting PI3K-Akt-mTOR Pathway on Lipid Metabolism Homeostasis in Goose Primary Hepatocytes. Anim. Int. J. Anim. Biosci. 2016, 10, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- González-Casanova, J.E.; Durán-Agüero, S.; Caro-Fuentes, N.J.; Gamboa-Arancibia, M.E.; Bruna, T.; Bermúdez, V.; Rojas-Gómez, D.M. New Insights on The Role of Connexins and Gap Junctions Channels in Adipose Tissue and Obesity. Int. J. Mol. Sci. 2021, 22, 12145. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, I.; Ferreira, C.; Moreira, D.; Kluck, G.E.G.; Barbosa, A.M.; Torrado, E.; Dinis-Oliveira, R.J.; Gonçalves, L.G.; Beauparlant, C.J.; Droit, A.; et al. The Absence of HIF-1 Increases Susceptibility to Leishmania Donovani Infection via Activation of BNIP3/mTOR/SREBP-1c Axis. Cell Rep. 2020, 30, 4052–4064. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, Z.; Zhao, C.; Wang, Y.; Wu, G.; Xiao, J.; McClain, C.J.; Li, X.; Feng, W. HIF-1α and HIF-2α are critically involved in hypoxia-induced lipid accumulation in hepatocytes through reducing PGC-1α-mediated fatty acid β-oxidation. Toxicol. Lett. 2014, 226, 117–123. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Number of FLNC Reads | Mean PID (%) | |
|---|---|---|---|
| Pre-Correction | Post-Correction | ||
| only pre-correction mapped | 9 | 51.86 | – |
| only post-correction mapped | 1075 | – | 86.79 |
| pre PID > post PID | 105,215 | 98.89 | 97.31 |
| pre PID = post PID | 372,471 | 99.38 | 99.38 |
| pre PID < post PID | 241,094 | 98.83 | 99.29 |
| Total | 719,864 | 98.97 | 99.03 |
| Feature | Pre-Correction | Post-Correction | Merged |
|---|---|---|---|
| unmapped | 1459 (0.20%) | 393 (0.05%) | 384 (0.05%) |
| multiple best mapped | 5881 (0.82%) | 6017 (0.84%) | 5633 (0.78%) |
| low-PID sequences | 17,250 (2.40%) | 27,294 (3.79%) | 14,551 (2.02%) |
| high-quality mapped | 695,658 (96.59%) | 686,544 (95.32%) | 699,680 (97.14%) |
| Feature | Loci of Reference Annotation | Loci of PacBio Annotation |
|---|---|---|
| Loci | 25,712 | 60,276 |
| Loci < 1 K | 5124 (19.93%) | 11,652 (19.33%) |
| Loci 1–2 K | 6032 (23.46%) | 22,074 (36.62%) |
| Loci 2–3 K | 4632 (18.01%) | 14,657 (24.32%) |
| Loci ≥ 3 K | 9924 (38.60%) | 11,893 (19.73%) |
| Total | 68,644 | 111,302 |
| Group | Node_Name | MCC | Gene_Name |
|---|---|---|---|
| SF vs. VF | NC_056057.1.1402.7 | 67 | COL1A2 |
| XM_004007726.5 | 67 | COL1A2 | |
| NC_056057.1.1402.33 | 59 | COL1A2 | |
| NC_056057.1.1402.18 | 57 | COL1A2 | |
| NC_056074.1.334.7 | 51 | EEF1G | |
| TF vs. SF | NC_056064.1.197.5 | 1.32 × 109 | PSMD11 |
| NC_056054.1.4780.11 | 1.32 × 109 | PSMD4 | |
| XM_015093596.3 | 1.32 × 109 | PSMD1 | |
| NC_056064.1.1711.2 | 1.32 × 109 | PSMD3 | |
| XM_012180724.3 | 1.32 × 109 | PSMC6 | |
| TF vs. VF | NC_056067.1.612.3 | 8.72 × 1013 | RPS19 |
| NC_056062.1.131.5 | 8.72 × 1013 | RPL8 | |
| NC_056077.1.54.1 | 8.72 × 1013 | RPS2 | |
| NC_056056.1.2665.21 | 8.72 × 1013 | RPL3 | |
| NC_056055.1.1036.4 | 8.72 × 1013 | RPS6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, L.; Pan, Y.; Yuan, M.; Wen, Z.; Qiao, L.; Wang, W.; Liu, J.; Li, B.; Liu, W. Full-Length Transcriptome and Gene Expression Analysis of Different Ovis aries Adipose Tissues Reveals Transcript Variants Involved in Lipid Biosynthesis. Animals 2024, 14, 7. https://doi.org/10.3390/ani14010007
An L, Pan Y, Yuan M, Wen Z, Qiao L, Wang W, Liu J, Li B, Liu W. Full-Length Transcriptome and Gene Expression Analysis of Different Ovis aries Adipose Tissues Reveals Transcript Variants Involved in Lipid Biosynthesis. Animals. 2024; 14(1):7. https://doi.org/10.3390/ani14010007
Chicago/Turabian StyleAn, Lixia, Yangyang Pan, Mengjiao Yuan, Zhonghao Wen, Liying Qiao, Weiwei Wang, Jianhua Liu, Baojun Li, and Wenzhong Liu. 2024. "Full-Length Transcriptome and Gene Expression Analysis of Different Ovis aries Adipose Tissues Reveals Transcript Variants Involved in Lipid Biosynthesis" Animals 14, no. 1: 7. https://doi.org/10.3390/ani14010007