
Investigation of ABO Gene Variants across More Than 60 Pig Breeds and Populations and Other Suidae Species Using Whole-Genome Sequencing Datasets
 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
- (i)
- ENA datasets (publicly available) were obtained from the sequencing of the genome of single animals [depth of sequencing (DP) > 5X over the whole genome and DP > 5X over the ABO gene and its 5 kb flanking regions]. This dataset was derived from 191 pigs from 20 European breeds, 105 pigs from 17 Asian breeds, 14 pigs from two breeds raised in South America, and two pig samples from one breed raised in New Zealand. Datasets from wild boars were also retrieved from the same database: two were from wild boars of the Near East region; 18 from wild boars of Asia; and 14 from wild boars of Europe. More details of this dataset and related sequencing-derived statistics are reported in Bovo et al. [16] and in Table S1;
- (ii)
- ENA datasets (publicly available) were obtained from a DNA pooling strategy (DP > 20X). This dataset consisted of 22 DNA pools (constructed from 30–35 pigs each), representing 22 different European breeds and one European wild boar population. More details of this dataset and related sequencing-derived statistics are reported in Bovo et al. [17,18] and in Table S1;
- (iii)
- Novel WGS datasets were produced from Italian Large White (no. = 90), Italian Duroc (no. = 35), and Italian Landrace (no. = 35) pigs. These pigs derived from triplets of siblings (two gilts and one castrated male) that were individually performance-tested at the central genetic station of the National Pig Breeders Association (ANAS) for the sib-testing evaluation of a boar from the same litter [19]. Animals were selected to be unrelated as much as possible based on pedigree analyses. Genomic DNA was extracted as described by Bovo et al. ([20]). Libraries with an average insert size of 350 bp were obtained and sequenced on the BGISeq 500 platform with a 150-bp long paired-end strategy, following the provider’s procedures. About 56 Gb were obtained from each pig, providing a DP of about 20X. More information on this dataset is reported in Table S1;
- (iv)
- ENA datasets from additional Sus species (Table S1) were included in the analyses, as these species are phylogenetically close to Sus scrofa [21]. These data were from Sus barbatus (no. = 3), Sus cebifrons (no. = 9), Sus celebensis (no. = 2), and Sus verrucosus (no. = 3). Since all extant Sus species, except Sus scrofa, are present in the Island Southeast Asia (ISEA) [21], we indicate them as ISEA species. Moreover, to obtain more informative phylogenetic analyses, we also included WGS datasets from two non-Sus species of the Suidae family (Table S1): Porcula salvania (no. = 6); and Phachochoerus africanus (no. = 15).
2.2. Sequencing Data Analysis and Detection of the 2.3 kb Insertion Allele
2.3. Sequencing Data Analysis and Detection of Other ABO Gene Region Variants
- (i)
- Average DP of the expanded ABO gene region (DPABO); this count was computed for alignments over both versions of the reference genome (Sscrofa11.1 and Sscrofa11.1+ABO);
- (ii)
- Average DP of the 39 bp sequence between the two deletion breakpoints (DPBP); this count was available only for the canonical Sscrofa11.1 reference genome;
- (iii)
- Average DP of the 2.3 kb deleted sequence (DPdel); this count was available only for the Sscrofa11.1+ABO genome version.
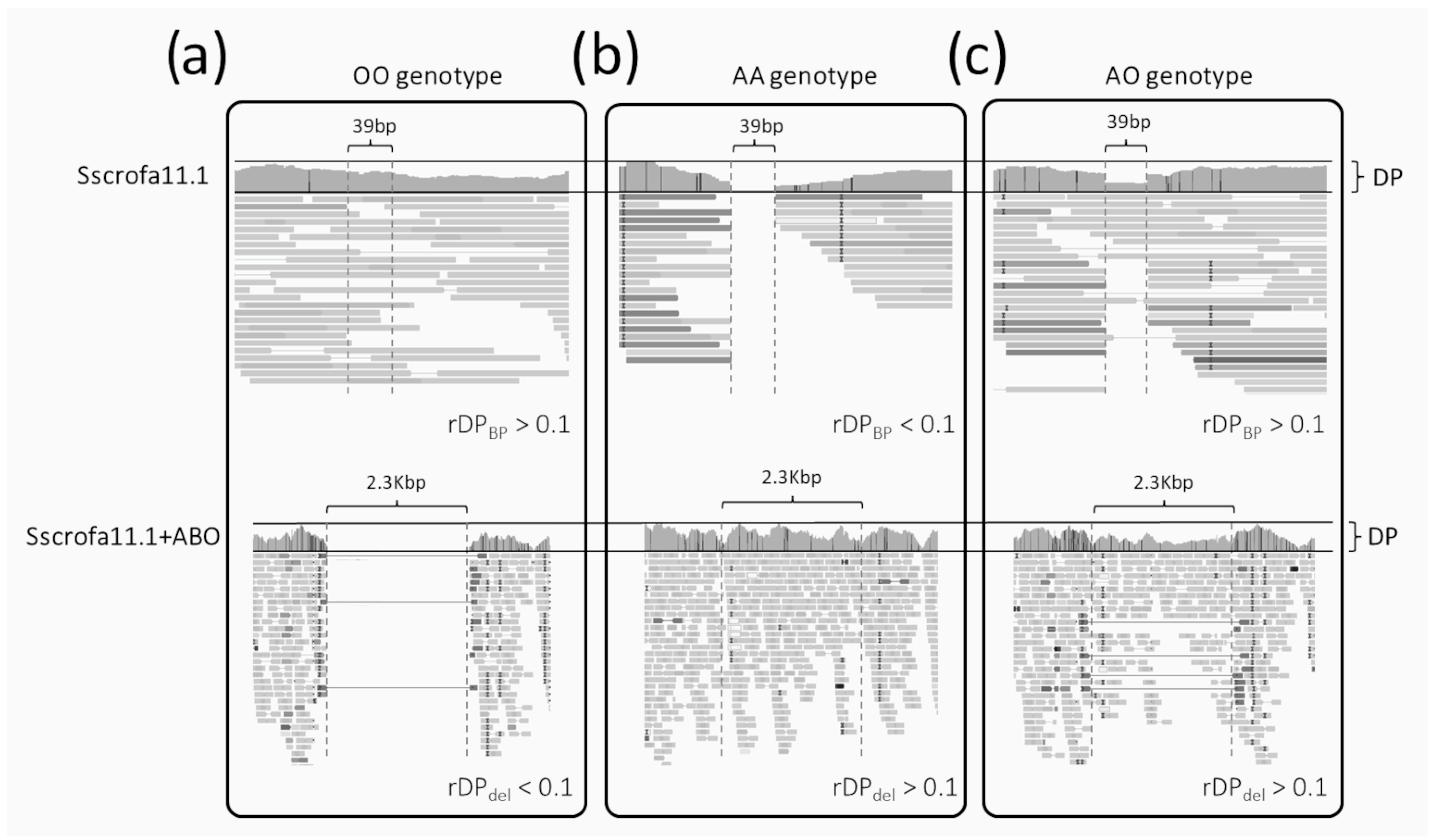
- (i)
- The homozygous OO genotype (i.e., deletion of the 2.3 kb sequence in both alleles; Figure 1a) is expected to have (i) reads covering the small region surrounded by the two breakpoints (DPBP > 0) when investigating the alignment over the canonical Sscrofa11.1 genome and (ii) no reads mapped over the 2.3 kb sequences when analyzing the alignments over the Sscrofa11.1+ABO genome version (DPdel~0). Considering ratios between counts and their simultaneous evaluation across the two reference genomes, genotype OO should have rDPdel < 0.1 and rDPBP > 0.1;
- (ii)
- The homozygous AA genotype (i.e., presence of the 2.3 kb sequence in both alleles; Figure 1b) is expected to have (i) no reads covering the small region surrounded by the two breakpoints (DPBP < 0.1; theoretically zero) when investigating the alignment over the canonical Sscrofa11.1 genome and (ii) reads mapped over the 2.3 kb sequences (with a similar average DP of the whole gene) when analyzing the alignment over the Sscrofa11.1+ABO genome version (DPdel > 0). Considering ratios between counts, genotype AA should have rDPdel > 0.1 and rDPBP < 0.1;
- (iii)
- The heterozygous AO genotype (Figure 1c) is expected to have (i) half of the reads fully covering the small region surrounded by the two breakpoints (DPBP > 0) and (ii) reads mapped over the 2.3 kb sequences (with half of the DP of the whole gene) when analyzing the alignment over the Sscrofa11.1+ABO genome version (DPdel > 0). Considering ratios between counts, the genotype AO should have rDPdel > 0.1 and rDPBP > 0.1.
2.4. Short and Structural Variant Discovery in the ABO Gene Region
2.5. Phylogenetic Analysis and Introgression Evaluation
2.6. Association Analyses with Production Traits and Blood Parameters in Italian Large White Pigs
3. Results
3.1. A/O In Silico Genotyping Results
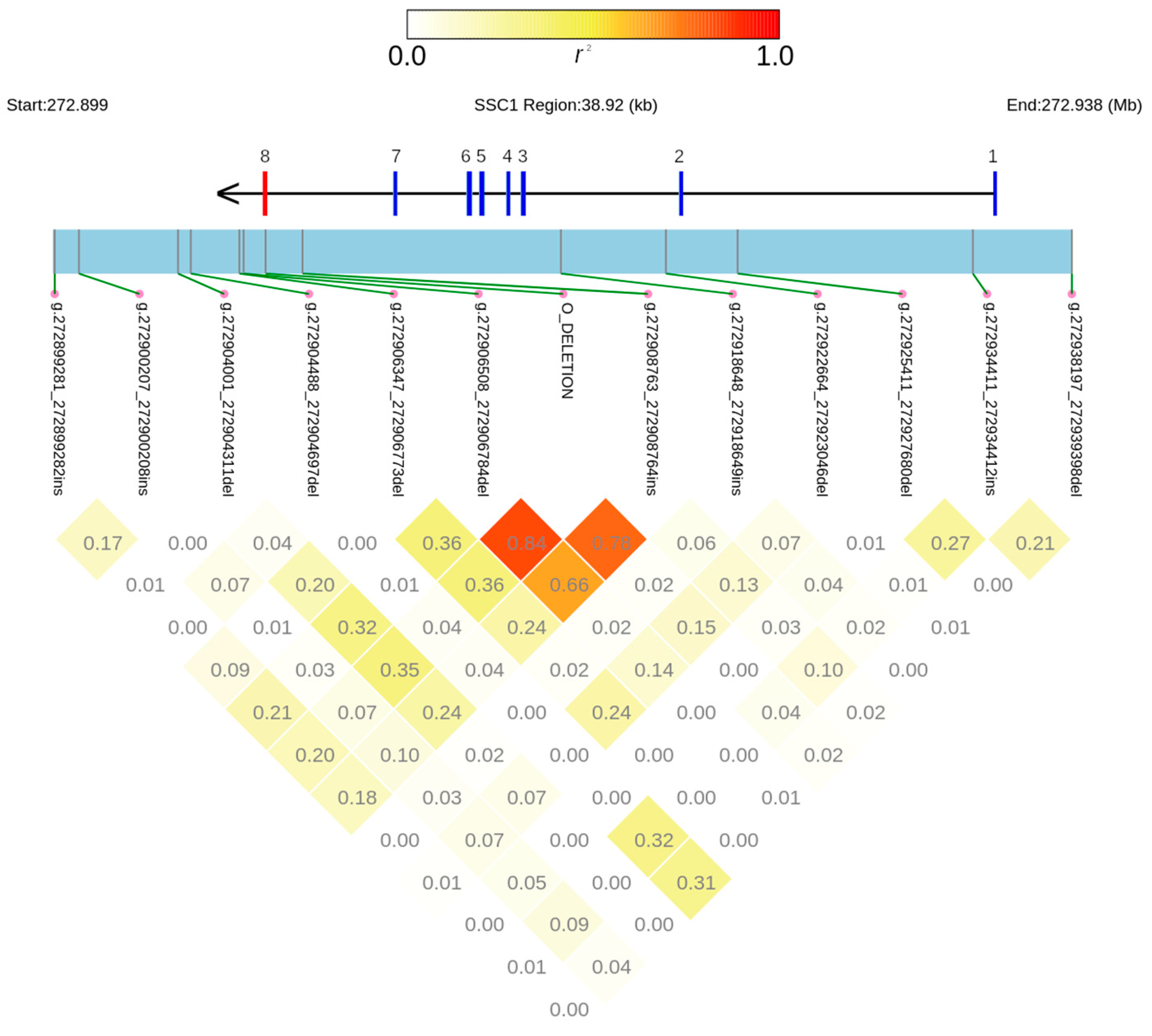
3.2. Other Structural Variants and Linkage Disequilibrium Study in the ABO Gene Region
3.3. ABO Gene Single Nucleotide Variants
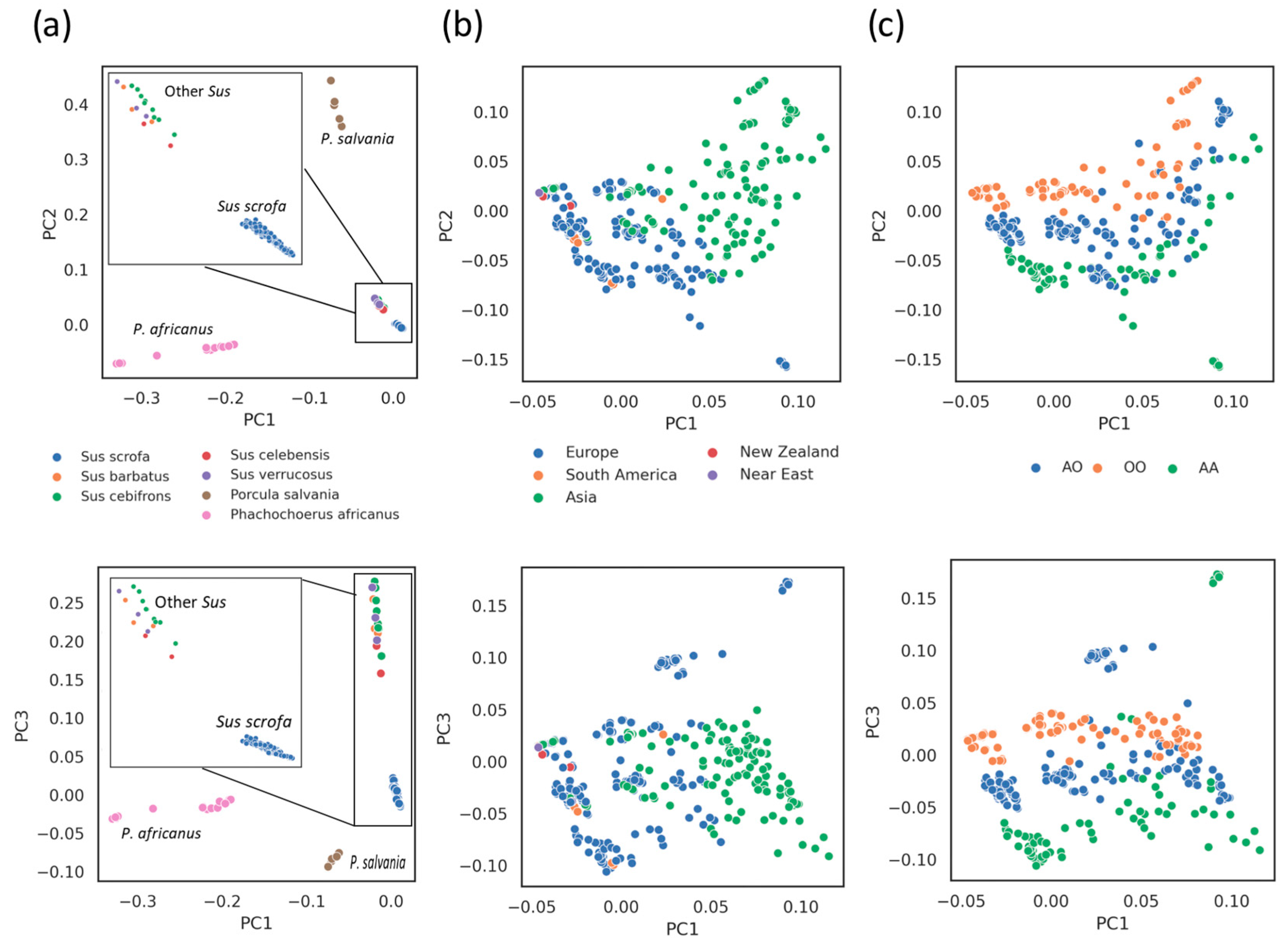
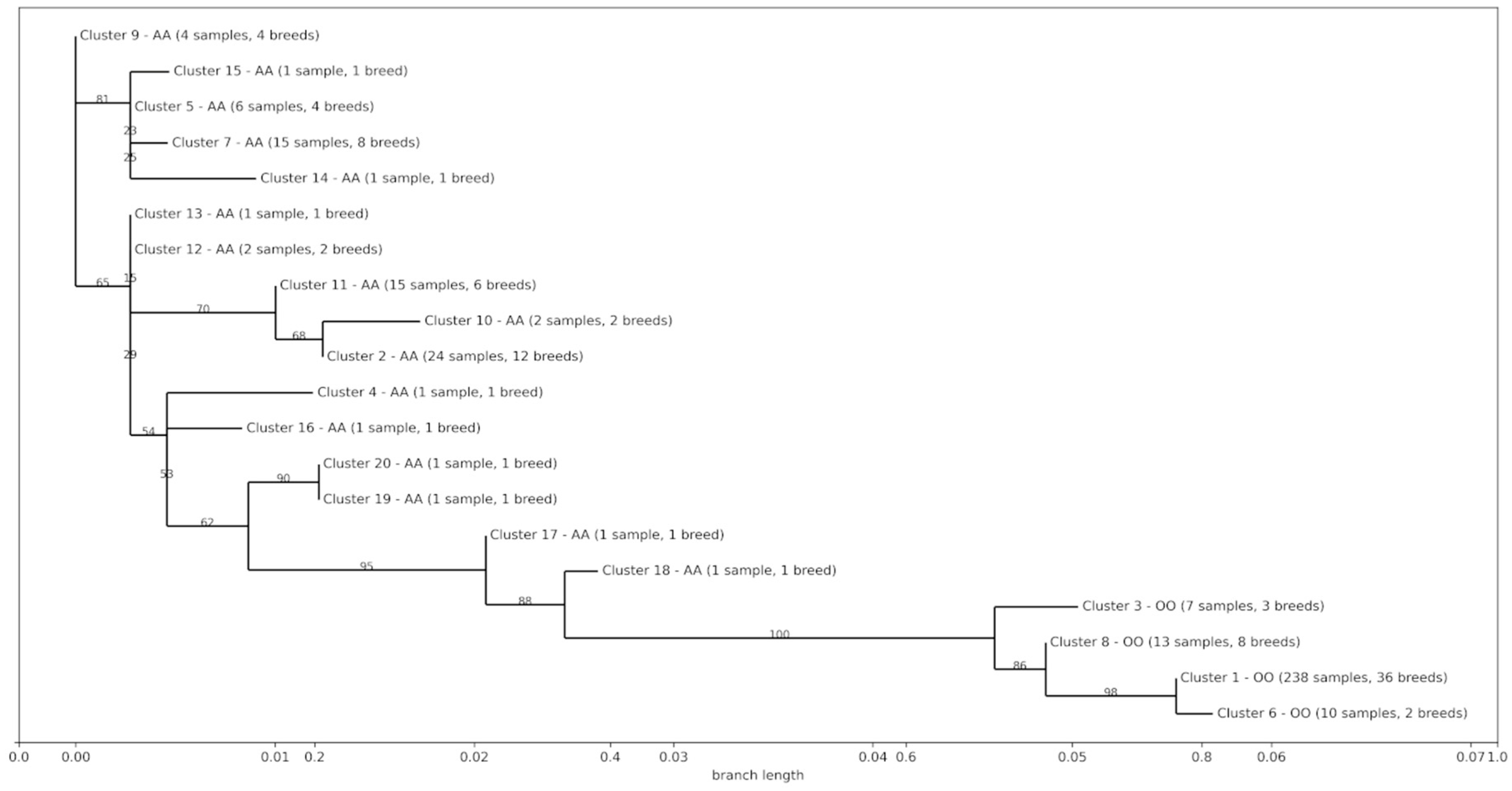
3.4. Phylogenetic and Introgression Analyses
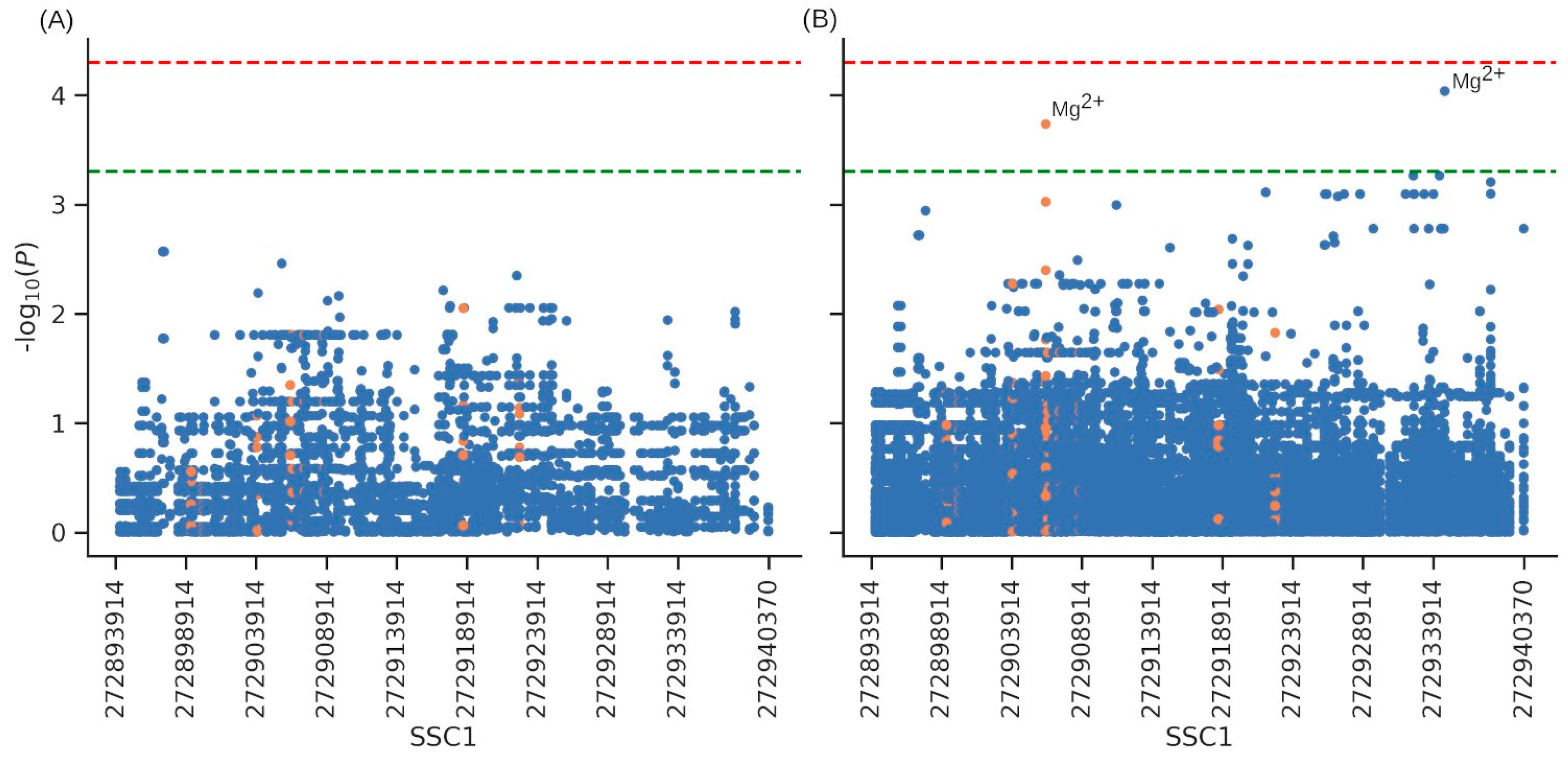
3.5. Association with Productive Traits and Haematological Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dabelsteen, E.; Gao, S. ABO Blood-Group Antigens in Oral Cancer. J. Dent. Res. 2005, 84, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, F.; Clausen, H.; White, T.; Marken, J.; Hakomori, S. Molecular Genetic Basis of the Histo-Blood Group ABO System. Nature 1990, 345, 229–233. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hong, X.; Zhang, J.; He, J.; Zhu, F.; Huang, H. Analysis of the Genomic Sequence of ABO Allele Using Next-Generation Sequencing Method. Front. Immunol. 2022, 13, 814263. [Google Scholar] [CrossRef] [PubMed]
- Storry, J.R. Nomenclature for Red Blood Cell Blood Group Alleles. Transfusion 2013, 53, 2844–2846. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.-F.; Zhu, F.; Li, X.-D.; An, Y.; Zhang, H.; Zhou, Y.; Zhang, X.; Jiang, Y. Association between ABO Gene Polymorphism (Rs505922) and Cancer Risk: A Meta-Analysis. Tumour Biol. 2015, 36, 5081–5087. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Huang, Y.; Zheng, H.; Li, L. Association of ABO Polymorphisms and Pancreatic Cancer/Cardiocerebrovascular Disease: A Meta-Analysis. BMC Med. Genet. 2020, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cai, Y.; Xu, A. Association Study of Polymorphisms in the ABO Gene and Their Gene-gene Interactions with Ischemic Stroke in Chinese Population. J. Clin. Lab. Anal. 2017, 32, e22329. [Google Scholar] [CrossRef]
- Shahbazi, S.; Mashayekhi, A.; Fatahi, N.; Mahdavi, M.-R. Association of ABO and Colton Blood Group Gene Polymorphisms With Hematological Traits Variation. Medicine 2015, 94, e2144. [Google Scholar] [CrossRef]
- Sinnott-Armstrong, N.; Tanigawa, Y.; Amar, D.; Mars, N.; Benner, C.; Aguirre, M.; Venkataraman, G.R.; Wainberg, M.; Ollila, H.M.; Kiiskinen, T.; et al. Genetics of 35 Blood and Urine Biomarkers in the UK Biobank. Nat. Genet. 2021, 53, 185–194. [Google Scholar] [CrossRef]
- Gustafsson, K.; Strahan, K.; Preece, A. A1,3Galactosyltransferase: A Target for in Vivo Genetic Manipulation in Xenotransplantation. Immunol. Rev. 1994, 141, 59–70. [Google Scholar] [CrossRef]
- Kominato, Y.; McNeill, P.D.; Yamamoto, M.; Russell, M.; Hakomori, S.; Yamamoto, F. Animal Histo-Blood Group ABO Genes. Biochem. Biophys. Res. Commun. 1992, 189, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Kermarrec, N.; Roubinet, F.; Apoil, P.A.; Blancher, A. Comparison of Allele O Sequences of the Human and Non-Human Primate ABO System. Immunogenetics 1999, 49, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, F.; Yamamoto, M. Molecular Genetic Basis of Porcine Histo-Blood Group AO System. Blood 2001, 97, 3308–3310. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wu, J.; Huang, X.; Zhou, Y.; Zhang, Y.; Liu, M.; Liu, Q.; Ke, S.; He, M.; Fu, H.; et al. ABO Genotype Alters the Gut Microbiota by Regulating GalNAc Levels in Pigs. Nature 2022, 606, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Ségurel, L.; Thompson, E.E.; Flutre, T.; Lovstad, J.; Venkat, A.; Margulis, S.W.; Moyse, J.; Ross, S.; Gamble, K.; Sella, G.; et al. The ABO Blood Group Is a Trans-Species Polymorphism in Primates. Proc. Natl. Acad. Sci. USA 2012, 109, 18493–18498. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Schiavo, G.; Bolner, M.; Ballan, M.; Fontanesi, L. Mining Livestock Genome Datasets for an Unconventional Characterization of Animal DNA Viromes. Genomics 2022, 114, 110312. [Google Scholar] [CrossRef]
- Bovo, S.; Ribani, A.; Muñoz, M.; Alves, E.; Araujo, J.P.; Bozzi, R.; Čandek-Potokar, M.; Charneca, R.; Di Palma, F.; Etherington, G.; et al. Whole-Genome Sequencing of European Autochthonous and Commercial Pig Breeds Allows the Detection of Signatures of Selection for Adaptation of Genetic Resources to Different Breeding and Production Systems. Genet. Sel. Evol. 2020, 52, 33. [Google Scholar] [CrossRef]
- Bovo, S.; Ribani, A.; Muñoz, M.; Alves, E.; Araujo, J.P.; Bozzi, R.; Charneca, R.; Di Palma, F.; Etherington, G.; Fernandez, A.I.; et al. Genome-Wide Detection of Copy Number Variants in European Autochthonous and Commercial Pig Breeds by Whole-Genome Sequencing of DNA Pools Identified Breed-Characterising Copy Number States. Anim. Genet. 2020, 51, 541–556. [Google Scholar] [CrossRef]
- Fontanesi, L.; Schiavo, G.; Galimberti, G.; Calò, D.G.; Scotti, E.; Martelli, P.L.; Buttazzoni, L.; Casadio, R.; Russo, V. A Genome Wide Association Study for Backfat Thickness in Italian Large White Pigs Highlights New Regions Affecting Fat Deposition Including Neuronal Genes. BMC Genom. 2012, 13, 583. [Google Scholar] [CrossRef]
- Bovo, S.; Schiavo, G.; Ribani, A.; Utzeri, V.J.; Taurisano, V.; Ballan, M.; Muñoz, M.; Alves, E.; Araujo, J.P.; Bozzi, R.; et al. Describing Variability in Pig Genes Involved in Coronavirus Infections for a One Health Perspective in Conservation of Animal Genetic Resources. Sci. Rep. 2021, 11, 3359. [Google Scholar] [CrossRef]
- Frantz, L.; Meijaard, E.; Gongora, J.; Haile, J.; Groenen, M.A.M.; Larson, G. The Evolution of Suidae. Annu. Rev. Anim. Biosci. 2016, 4, 61–85. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Picard Tools—By Broad Institute. Available online: https://broadinstitute.github.io/picard/ (accessed on 11 October 2023).
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick Coverage Calculation for Genomes and Exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural Variant Discovery by Integrated Paired-End and Split-Read Analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Pig [Sus Scrofa] Genomic Dataset. Available online: https://www.repeatmasker.org/species/susScr.html (accessed on 11 October 2023).
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Dong, S.-S.; He, W.-M.; Ji, J.-J.; Zhang, C.; Guo, Y.; Yang, T.-L. LDBlockShow: A Fast and Convenient Tool for Visualizing Linkage Disequilibrium and Haplotype Blocks Based on Variant Call Format Files. Brief. Bioinform. 2021, 22, bbaa227. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Tian, X.; Zhou, Y.; Browning, S.R. Fast Two-Stage Phasing of Large-Scale Sequence Data. Am. J. Hum. Genet. 2021, 108, 1880–1890. [Google Scholar] [CrossRef] [PubMed]
- Tortereau, F.; Servin, B.; Frantz, L.; Megens, H.-J.; Milan, D.; Rohrer, G.; Wiedmann, R.; Beever, J.; Archibald, A.L.; Schook, L.B.; et al. A High Density Recombination Map of the Pig Reveals a Correlation between Sex-Specific Recombination and GC Content. BMC Genom. 2012, 13, 586. [Google Scholar] [CrossRef] [PubMed]
- Nassar, L.R.; Barber, G.P.; Benet-Pagès, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, B.T.; et al. The UCSC Genome Browser Database: 2023 Update. Nucleic Acids Res. 2023, 51, D1188–D1195. [Google Scholar] [CrossRef]
- Browning, B.L.; Browning, S.R. Improving the Accuracy and Efficiency of Identity-by-Descent Detection in Population Data. Genetics 2013, 194, 459–471. [Google Scholar] [CrossRef]
- Malinsky, M.; Matschiner, M.; Svardal, H. Dsuite—Fast D-Statistics and Related Admixture Evidence from VCF Files. Mol. Ecol. Resour. 2021, 21, 584–595. [Google Scholar] [CrossRef]
- Zheng, Y.; Janke, A. Gene Flow Analysis Method, the D-Statistic, Is Robust in a Wide Parameter Space. BMC Bioinform. 2018, 19, 10. [Google Scholar] [CrossRef]
- Fontanesi, L.; Bertolini, F.; Scotti, E.; Trevisi, P.; Buttazzoni, L.; Dall’olio, S.; Davoli, R.; Bosi, P.; Russo, V. Polymorphisms in an Obesity-Related Gene (PCSK1) Are Associated with Fat Deposition and Production Traits in Italian Heavy Pigs. Animal 2012, 6, 1913–1924. [Google Scholar] [CrossRef]
- Bovo, S.; Schiavo, G.; Mazzoni, G.; Dall’Olio, S.; Galimberti, G.; Calò, D.G.; Scotti, E.; Bertolini, F.; Buttazzoni, L.; Samorè, A.B.; et al. Genome-Wide Association Study for the Level of Serum Electrolytes in Italian Large White Pigs. Anim. Genet. 2016, 47, 597–602. [Google Scholar] [CrossRef]
- Bovo, S.; Mazzoni, G.; Bertolini, F.; Schiavo, G.; Galimberti, G.; Gallo, M.; Dall’Olio, S.; Fontanesi, L. Genome-Wide Association Studies for 30 Haematological and Blood Clinical-Biochemical Traits in Large White Pigs Reveal Genomic Regions Affecting Intermediate Phenotypes. Sci. Rep. 2019, 9, 7003. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Efficient Multivariate Linear Mixed Model Algorithms for Genome-Wide Association Studies. Nat. Methods 2014, 11, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Genome-Wide Association Study of 14,000 Cases of Seven Common Diseases and 3000 Shared Controls. Nature 2007, 447, 661–678. [Google Scholar]
- Bertolini, F.; Schiavo, G.; Galimberti, G.; Bovo, S.; D’Andrea, M.; Gallo, M.; Buttazzoni, L.; Rothschild, M.F.; Fontanesi, L. Genome-Wide Association Studies for Seven Production Traits Highlight Genomic Regions Useful to Dissect Dry-Cured Ham Quality and Production Traits in Duroc Heavy Pigs. Animal 2018, 12, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.-P.; Tribout, T.; Iannuccelli, N.; Bouffaud, M.; Servin, B.; Tenghe, A.; Dehais, P.; Muller, N.; Del Schneider, M.P.; Mercat, M.-J.; et al. A Genome-Wide Association Study of Production Traits in a Commercial Population of Large White Pigs: Evidence of Haplotypes Affecting Meat Quality. Genet. Sel. Evol. 2014, 46, 12. [Google Scholar] [CrossRef]
- Cooper, D.K.C.; Gaston, R.; Eckhoff, D.; Ladowski, J.; Yamamoto, T.; Wang, L.; Iwase, H.; Hara, H.; Tector, M.; Tector, A.J. Xenotransplantation—The Current Status and Prospects. Br. Med. Bull. 2018, 125, 5–14. [Google Scholar] [CrossRef]
- Choi, M.-K.; Le, M.T.; Cho, H.; Yum, J.; Kang, M.; Song, H.; Kim, J.-H.; Chung, H.J.; Hong, K.; Park, C. Determination of Complete Sequence Information of the Human ABO Blood Group Orthologous Gene in Pigs and Breed Difference in Blood Type Frequencies. Gene 2018, 640, 1–5. [Google Scholar] [CrossRef]
- Groenen, M.A.M.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.-J.; et al. Analyses of Pig Genomes Provide Insight into Porcine Demography and Evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef]
- Liu, L.; Bosse, M.; Megens, H.-J.; Frantz, L.A.F.; Lee, Y.-L.; Irving-Pease, E.K.; Narayan, G.; Groenen, M.A.M.; Madsen, O. Genomic Analysis on Pygmy Hog Reveals Extensive Interbreeding during Wild Boar Expansion. Nat. Commun. 2019, 10, 1992. [Google Scholar] [CrossRef]
- Gieger, C.; Geistlinger, L.; Altmaier, E.; de Angelis, M.H.; Kronenberg, F.; Meitinger, T.; Mewes, H.-W.; Wichmann, H.-E.; Weinberger, K.M.; Adamski, J.; et al. Genetics Meets Metabolomics: A Genome-Wide Association Study of Metabolite Profiles in Human Serum. PLoS Genet. 2008, 4, e1000282. [Google Scholar] [CrossRef]
- Bovo, S.; Mazzoni, G.; Galimberti, G.; Calò, D.G.; Fanelli, F.; Mezzullo, M.; Schiavo, G.; Manisi, A.; Trevisi, P.; Bosi, P.; et al. Metabolomics Evidences Plasma and Serum Biomarkers Differentiating Two Heavy Pig Breeds. Animal 2016, 10, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Reiner, G.; Clemens, N.; Lohner, E.; Willems, H. SNPs in the Porcine GOT1 Gene Improve a QTL for Serum Aspartate Aminotransferase Activity on SSC14. Anim. Genet. 2010, 41, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.-K.; Cho, I.-C.; Lee, J.-B.; Jung, E.-J.; Lim, H.-T.; Han, S.-H.; Lee, S.-S.; Ko, M.-S.; Kang, T.; Hwang, J.-H.; et al. QTL Analysis of Clinical-Chemical Traits in an F2 Intercross between Landrace and Korean Native Pigs. Physiol. Genom. 2012, 44, 657–668. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, D.N.; Warner, R.D.; Leury, B.J.; Dunshea, F.R. The Effect of Dietary Magnesium Aspartate Supplementation on Pork Quality. J. Anim. Sci. 1998, 76, 104–109. [Google Scholar] [CrossRef]
- Lipiński, K.; Zuk-Golaszewska, K.; Purwin, C.; Stasiewicz, M. Effects of Magnesium on Pork Quality. J. Elem. 2011, 16, 325–337. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed/Species | Origin | No. of Pig | Genotypes 1 | Alleles 2 | |||
|---|---|---|---|---|---|---|---|
| OO | AO | AA | O | A | |||
| Sus scrofa (total) | 506 | 260 | 175 | 71 | 0.69 | 0.31 | |
| Sus scrofa (Asian only) | |||||||
| Anqing Six-end-white pig | Asia | 18 | 3 | 12 | 3 | 0.50 | 0.50 |
| Asian wild boar | Asia | 18 | 4 | 9 | 5 | 0.47 | 0.53 |
| Erhualian | Asia | 13 | 8 | 4 | 1 | 0.77 | 0.23 |
| Jeju black pig | Asia | 8 | 5 | 3 | 0 | 0.81 | 0.19 |
| Korean pig | Asia | 6 | 6 | 0 | 0 | 1.00 | 0.00 |
| Meishan | Asia | 34 | 14 | 15 | 5 | 0.63 | 0.37 |
| Rongchang | Asia | 5 | 1 | 2 | 2 | 0.40 | 0.60 |
| Sus scrofa (European only) | |||||||
| Duroc | Europe | 36 | 11 | 18 | 7 | 0.56 | 0.44 |
| European wild boar | Europe | 14 | 1 | 5 | 8 | 0.25 | 0.75 |
| Goettingen minipig | Europe | 8 | 0 | 5 | 3 | 0.31 | 0.69 |
| Italian Duroc | Europe | 35 | 9 | 24 | 2 | 0.60 | 0.40 |
| Italian Landrace | Europe | 35 | 31 | 4 | 0 | 0.94 | 0.06 |
| Italian Large White | Europe | 90 | 67 | 20 | 3 | 0.86 | 0.14 |
| Landrace | Europe | 19 | 15 | 4 | 0 | 0.89 | 0.11 |
| Large White | Europe | 63 | 40 | 16 | 7 | 0.76 | 0.24 |
| Mangalica | Europe | 5 | 2 | 3 | 0 | 0.70 | 0.30 |
| Pietrain | Europe | 17 | 7 | 5 | 5 | 0.56 | 0.44 |
| Yorkshire | Europe | 18 | 4 | 6 | 8 | 0.39 | 0.61 |
| Yucatan miniature pig | South America | 13 | 4 | 7 | 2 | 0.58 | 0.42 |
| Sus cebifrons | - | 9 | 5 | 4 | 0 | 0.78 | 0.22 |
| Porcula salvania | - | 6 | 0 | 0 | 6 | 0.00 | 1.00 |
| Phachochoerus africanus | - | 15 | 0 | 0 | 15 | 0.00 | 1.00 |
| Breed/Population | Allele O | Allele A |
|---|---|---|
| Alentejana | 0.51 | 0.49 |
| Apulo Calabrese | 0.72 | 0.28 |
| Basque | 0.47 | 0.53 |
| Black Slavonian | 0.53 | 0.47 |
| Bísara | 0.46 | 0.54 |
| Casertana | 0.52 | 0.48 |
| Cinta Senese | 0.85 | 0.15 |
| European Wild Boar | 0.35 | 0.65 |
| Gascon | 0.47 | 0.53 |
| Italian Duroc | 0.58 | 0.42 |
| Italian Landrace | 0.93 | 0.07 |
| Italian Large White | 0.80 | 0.20 |
| Krškopolje | 0.48 | 0.52 |
| Lithuanian White Old type | 0.84 | 0.16 |
| Lithuanian Indigenous Wattle | 0.77 | 0.23 |
| Majorcan Black | 0.37 | 0.63 |
| Mora Romagnola | 0.21 | 0.79 |
| Moravka | 0.69 | 0.31 |
| Nero Siciliano | 0.65 | 0.35 |
| Sarda | 0.51 | 0.49 |
| Schwäbisch Hällisches Schwein | 0.78 | 0.22 |
| Swallow-Bellied Mangalitsa | 0.29 | 0.71 |
| Turopolje | 0.61 | 0.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolner, M.; Bertolini, F.; Bovo, S.; Schiavo, G.; Fontanesi, L. Investigation of ABO Gene Variants across More Than 60 Pig Breeds and Populations and Other Suidae Species Using Whole-Genome Sequencing Datasets. Animals 2024, 14, 5. https://doi.org/10.3390/ani14010005
Bolner M, Bertolini F, Bovo S, Schiavo G, Fontanesi L. Investigation of ABO Gene Variants across More Than 60 Pig Breeds and Populations and Other Suidae Species Using Whole-Genome Sequencing Datasets. Animals. 2024; 14(1):5. https://doi.org/10.3390/ani14010005
Chicago/Turabian StyleBolner, Matteo, Francesca Bertolini, Samuele Bovo, Giuseppina Schiavo, and Luca Fontanesi. 2024. "Investigation of ABO Gene Variants across More Than 60 Pig Breeds and Populations and Other Suidae Species Using Whole-Genome Sequencing Datasets" Animals 14, no. 1: 5. https://doi.org/10.3390/ani14010005
APA StyleBolner, M., Bertolini, F., Bovo, S., Schiavo, G., & Fontanesi, L. (2024). Investigation of ABO Gene Variants across More Than 60 Pig Breeds and Populations and Other Suidae Species Using Whole-Genome Sequencing Datasets. Animals, 14(1), 5. https://doi.org/10.3390/ani14010005