
Dynamic Changes in Intestinal Gene Expression and Microbiota across Chicken Egg-Laying Stages
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Transcriptome Sequence and Data Analysis
2.3. Microbial DNA Extraction and Microbiome Sequencing
2.4. Ethical Approval
2.5. Data Availability
3. Results
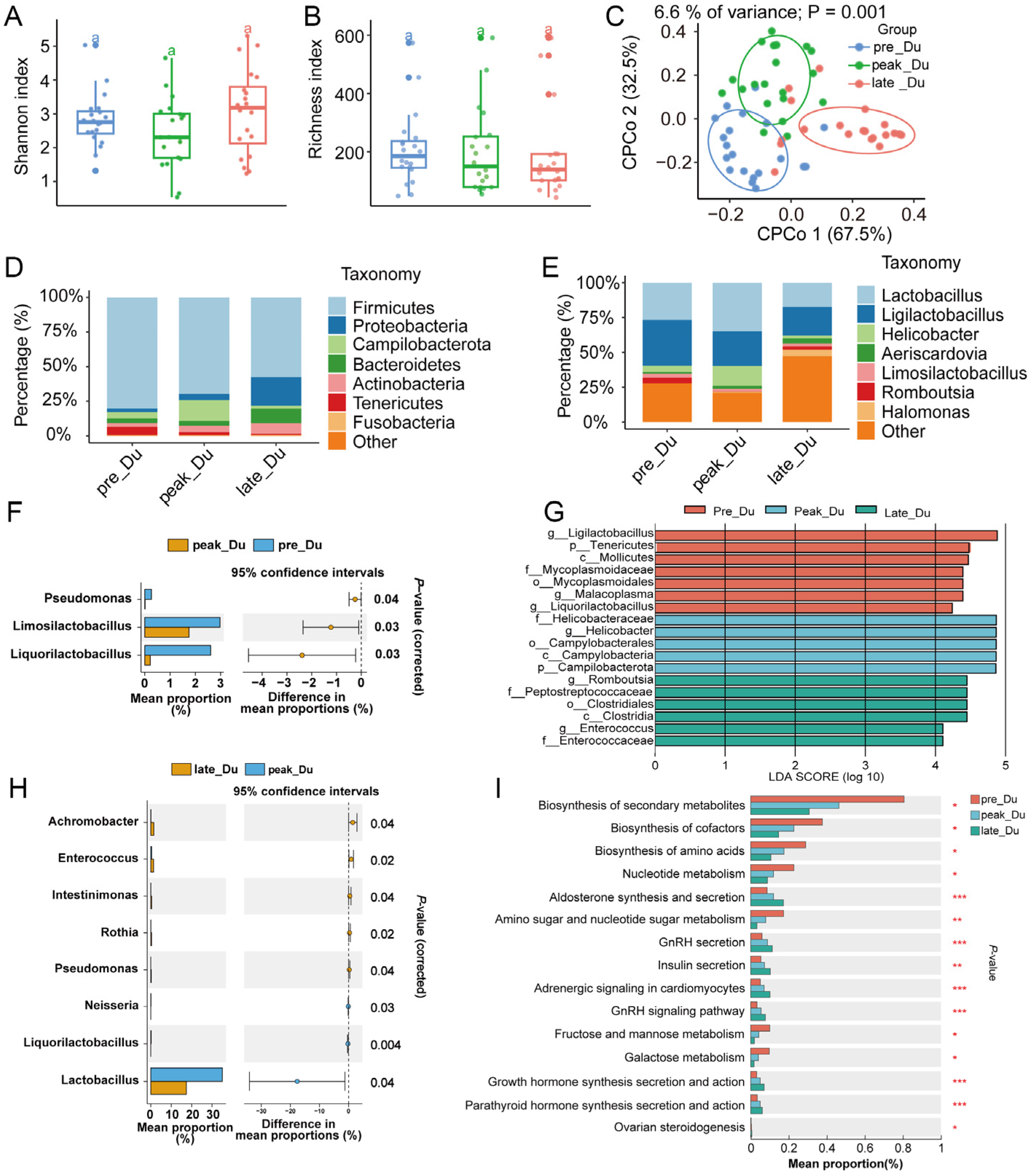
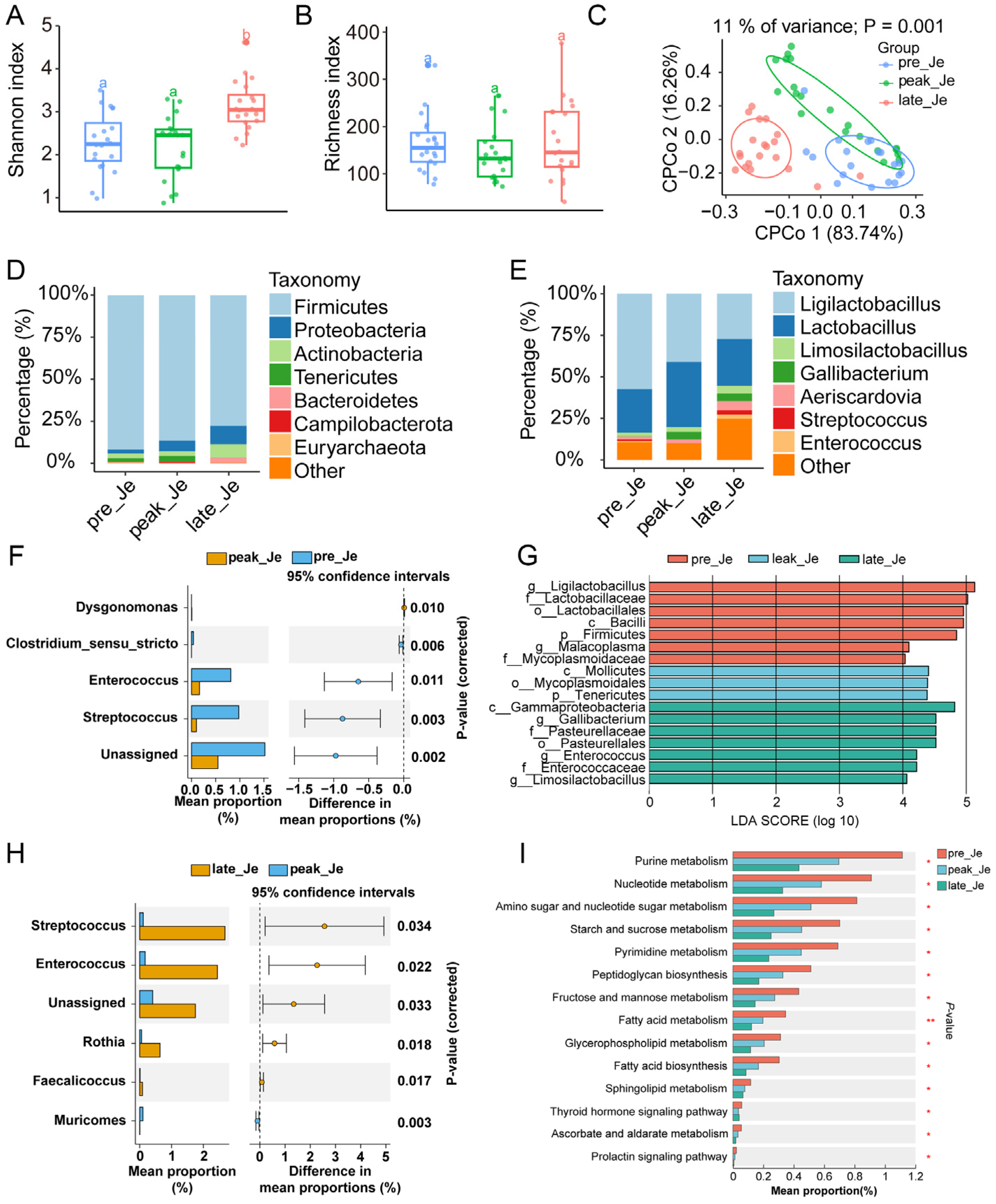
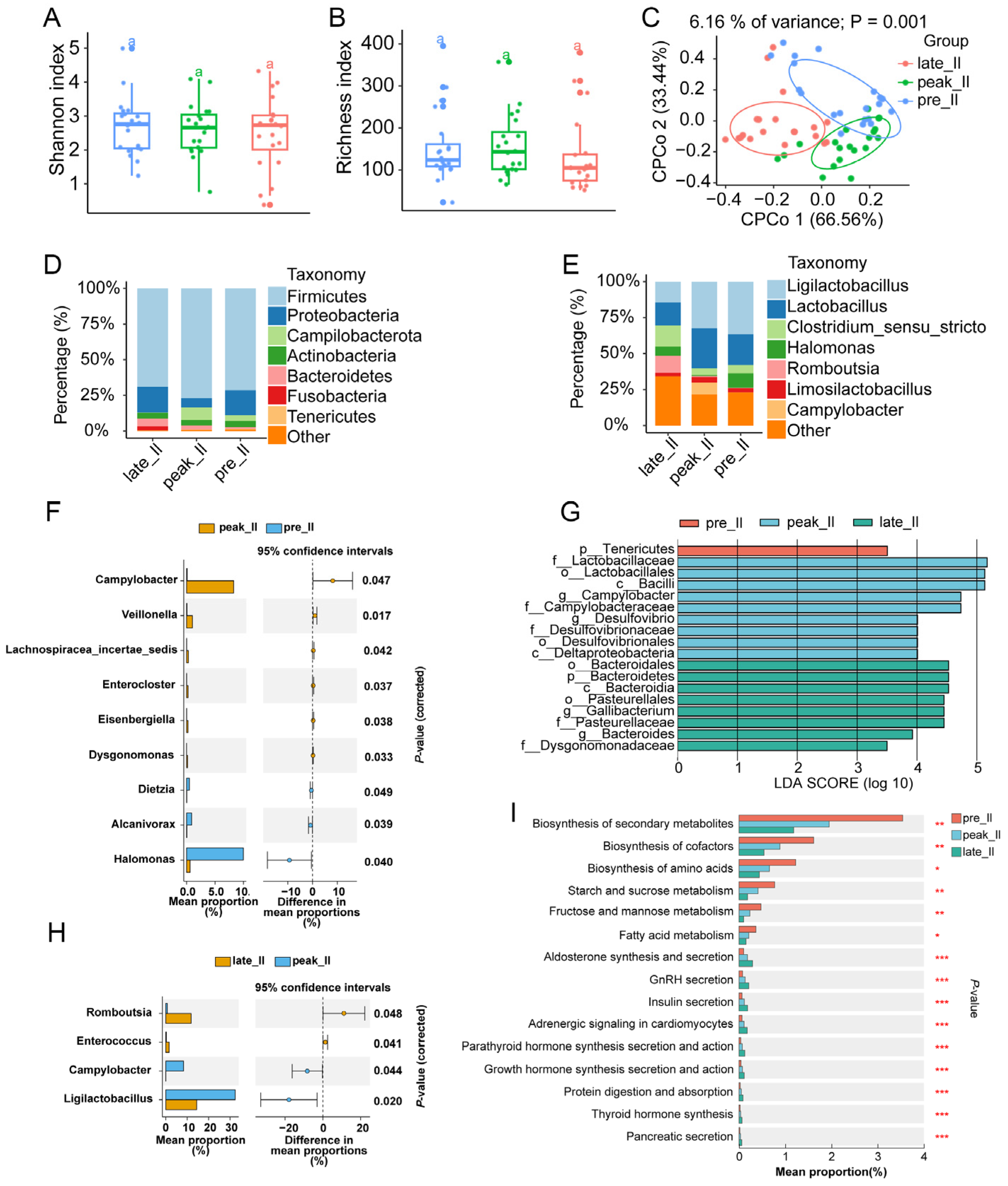
3.1. 16s rRNA Sequencing Information of the Duodenum, Jejunum, and Ileum Content during Egg-Laying Periods
3.2. Dynamically Microbial Changes in Duodenum during Egg-Laying Periods
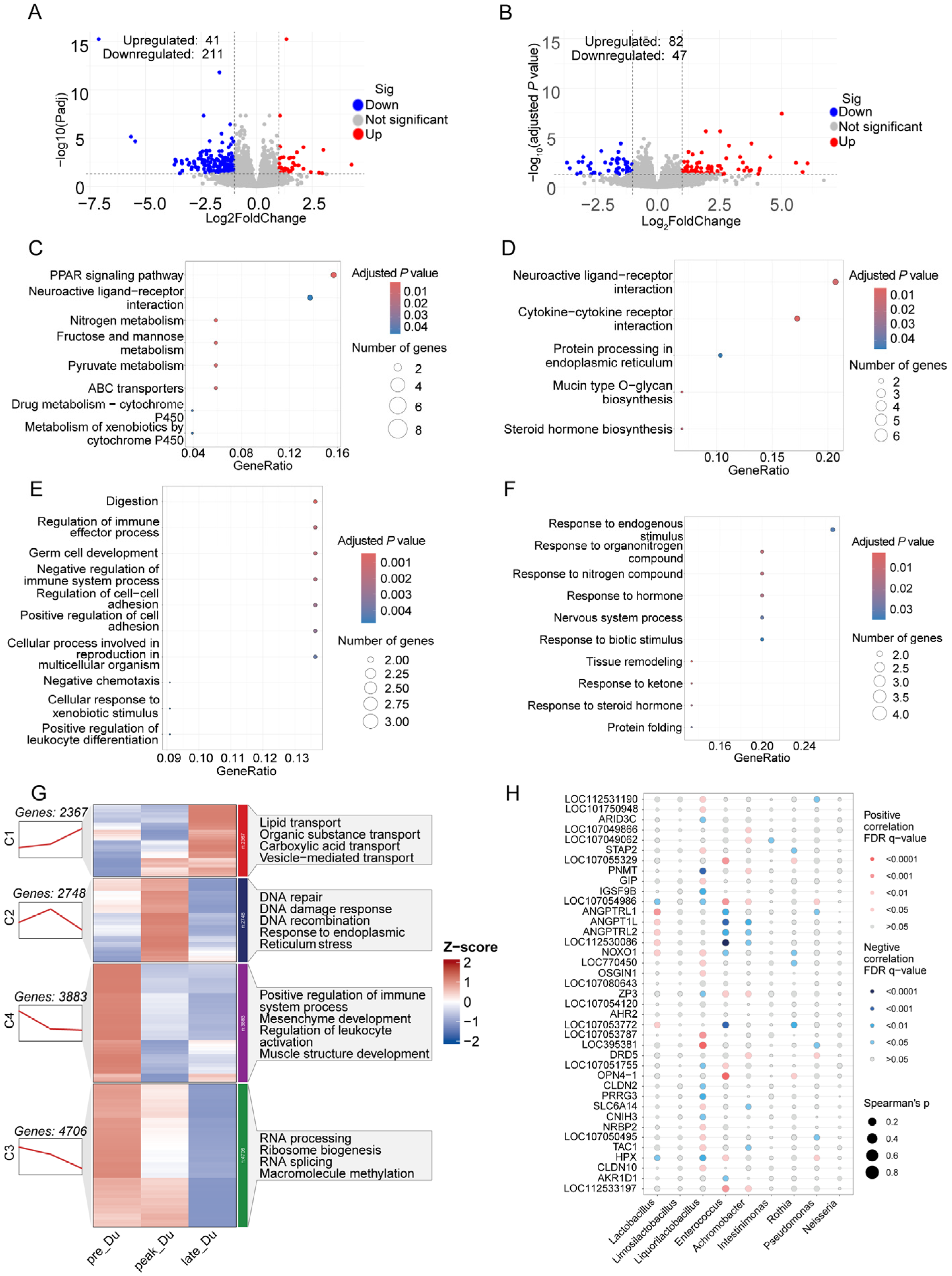
3.3. Transcriptome Analysis of Duodenum during Egg-Laying Periods
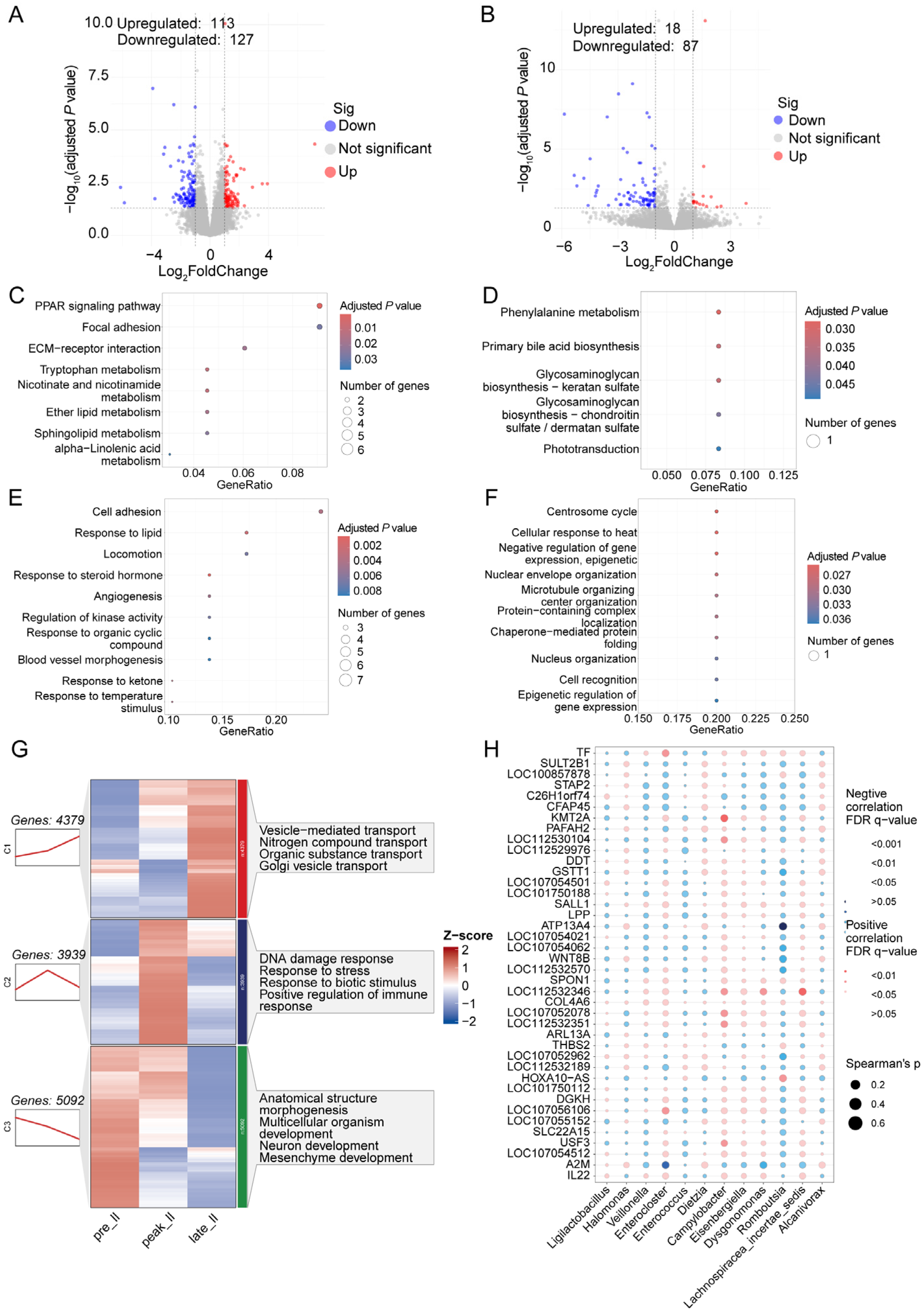
3.4. Dynamically Microbial Changes in Jejunum during Egg-Laying Periods
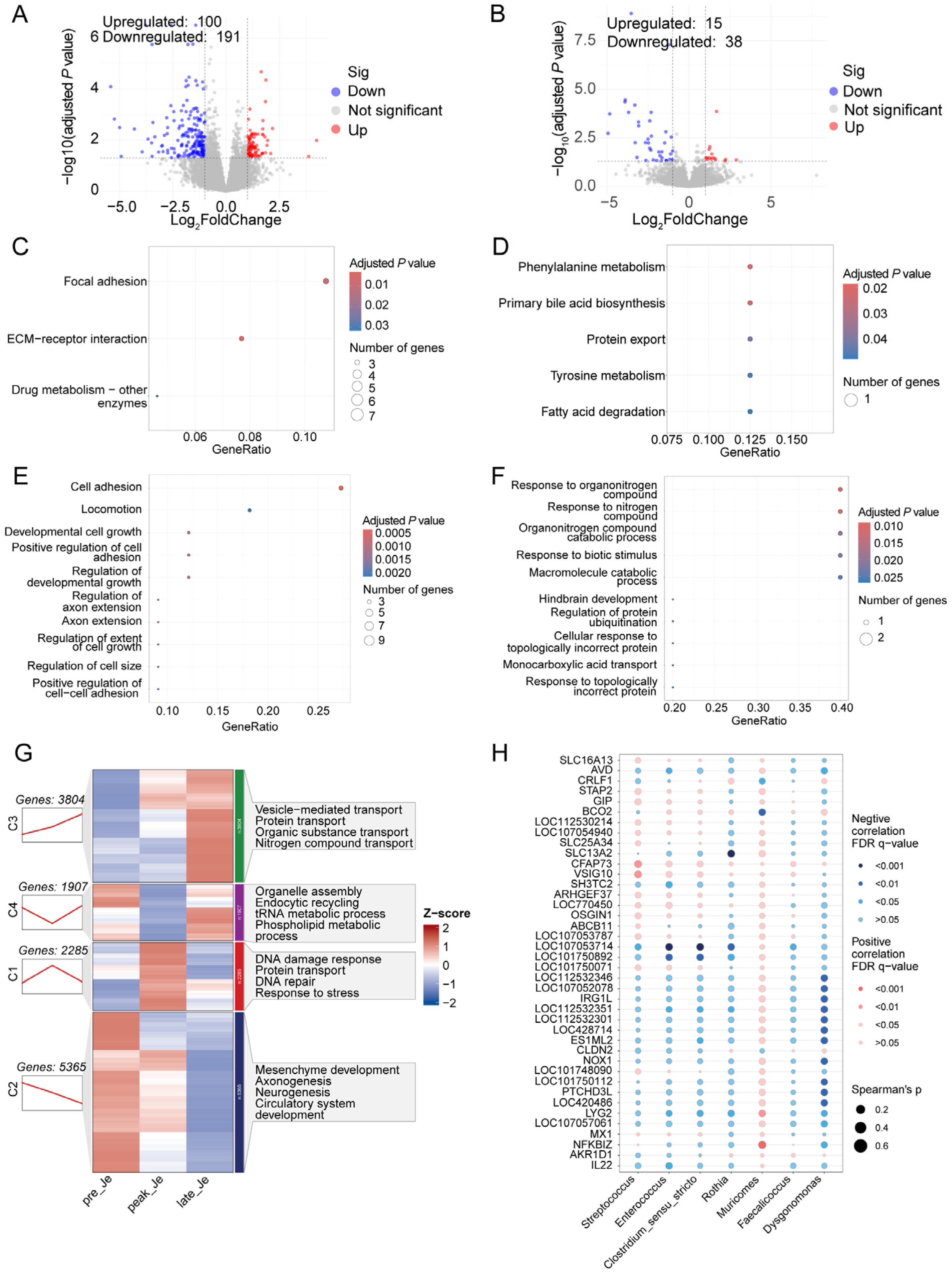
3.5. Transcriptome Analysis of Jejunum during Egg-Laying Periods
3.6. Dynamically Microbial Changes in Ileum during Egg-Laying Periods
3.7. Transcriptome Analysis of Ileum during Egg-Laying Periods
4. Discussion
4.1. Changes in Duodenal Microbiota and Gene Expression during Egg Laying
4.2. Changed Microbiota and Gene Expression in Jejunum during Egg Laying
4.3. Changes in Ileal Microbiota and Gene Expression during Egg Laying
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, H.; Li, H.; Hou, Y.; Huang, L.; Hu, J.; Lu, Y.; Liu, X. Differences in egg yolk precursor formation of Guangxi Ma chickens with dissimilar laying rate at the same or various ages. Theriogenology 2022, 184, 13–25. [Google Scholar] [CrossRef]
- Ritzi, M.M.; Abdelrahman, W.; Mohnl, M.; Dalloul, R.A. Effects of probiotics and application methods on performance and response of broiler chickens to an Eimeria challenge. Poult. Sci. 2014, 93, 2772–2778. [Google Scholar] [CrossRef]
- Nii, T.; Bungo, T.; Isobe, N.; Yoshimura, Y. Slight disruption in intestinal environment by dextran sodium sulfate reduces egg yolk size through disfunction of ovarian follicle growth. Front. Physiol. 2020, 11, 607369. [Google Scholar] [CrossRef]
- Su, Y.; Ge, Y.; Xu, Z.; Zhang, D.; Li, D. The digestive and reproductive tract microbiotas and their association with body weight in laying hens. Poult. Sci. 2021, 100, 101422. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Tian, S.; Li, D.; Zhu, W.; Wang, T.; Mishra, S.K.; Wei, R.; Xu, Z.; He, M.; Zhao, X.; et al. Association of female reproductive tract microbiota with egg production in layer chickens. Gigascience 2021, 10, giab067. [Google Scholar] [CrossRef]
- Joat, N.; Van, T.T.H.; Stanley, D.; Moore, R.J.; Chousalkar, K. Temporal dynamics of gut microbiota in caged laying hens: A field observation from hatching to end of lay. Appl. Microbiol. Biotechnol. 2021, 105, 4719–4730. [Google Scholar] [CrossRef]
- Lumpkins, B.S.; Batal, A.B.; Lee, M. The effect of gender on the bacterial community in the gastrointestinal tract of broilers. Poult. Sci. 2008, 87, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, L.; Sun, X.; Wan, X.; Sun, G.; Jiang, R.; Li, W.; Tian, Y.; Liu, X.; Kang, X. Characteristics of the fecal microbiota of high- and low-yield hens and effects of fecal microbiota transplantation on egg production performance. Res. Vet. Sci. 2020, 129, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Videnska, P.; Sedlar, K.; Lukac, M.; Faldynova, M.; Gerzova, L.; Cejkova, D.; Sisak, F.; Rychlik, I. Succession and replacement of bacterial populations in the caecum of egg laying hens over their whole life. PLoS ONE 2014, 9, e115142. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Gu, T.; Xu, Q.; Zhu, G.; Chen, G. Effects of Salmonella enterica serovar enteritidis infection on egg production and the immune response of the laying duck Anas platyrhynchos. PeerJ 2019, 7, e6359. [Google Scholar] [CrossRef]
- Wang, C.-L.; Fan, Y.-C.; Chun-Hsien, T.; Chiu, C.-H.; Tsai, H.-J.; Chou, C.-H. Salmonella enteritidis infection slows steroidogenesis and impedes cell growth in hen granulosa Cells. Avian Dis. 2014, 58, 511–517. [Google Scholar] [CrossRef]
- Wang, W.; Wang, J.; Zhang, H.; Wu, S.; Qi, G. Effects of Clostridium butyricum on production performance and intestinal absorption function of laying hens in the late phase of production. Anim. Feed Sci. Technol. 2020, 264, 114476. [Google Scholar] [CrossRef]
- Furuse, M.; Okumura, J. Nutritional and physiological characteristics in germ-free chickens. Comp. Biochem. Physiol. A Physiol. 1994, 109, 547–556. [Google Scholar] [PubMed]
- Gabriel, I.; Lessire, M.; Mallet, S.; Guillot, J.F. Microflora of the digestive tract: Critical factors and consequences for poultry. World’s Poult. Sci. J. 2006, 62, 499–511. [Google Scholar] [CrossRef]
- Zhang, H.; Ding, X.; Bai, S.; Zeng, Q.; Zhang, K.; Mao, X.; Chu, L.; Hou, D.; Xuan, Y.; Wang, J. Alleviating effect of dietary supplementation of benzoic acid, Enterococcus Faecium and essential oil complex on coccidia and Clostridium perfringens challenge in laying hens. Poult. Sci. 2022, 101, 101720. [Google Scholar] [CrossRef]
- Beldowska, A.; Barszcz, M.; Dunislawska, A. State of the art in research on the gut-liver and gut-brain axis in poultry. J. Anim. Sci. Biotechnol. 2023, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Mattner, J. Impact of microbes on the pathogenesis of primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC). Int. J. Mol. Sci. 2016, 17, 1864. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Ding, Q.; Zhang, W.; Kang, M.; Ma, J.; Zhao, L. Gut microbial beta-glucuronidase: A vital regulator in female estrogen metabolism. Gut Microbes 2023, 15, 2236749. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Tian, Y.; Hou, M.; Zhao, Y.; Li, J.; Aftab, U.; Rousseau, X.; Jiang, R.; Kang, X.; Tian, Y.; et al. Evaluation of stimbiotic on growth performance and intestinal development of broilers fed corn- or wheat-based diets. Poult. Sci. 2023, 102, 103094. [Google Scholar] [CrossRef]
- Shao, M.; Shi, K.; Zhao, Q.; Duan, Y.; Shen, Y.; Tian, J.; He, K.; Li, D.; Yu, M.; Lu, Y.; et al. Transcriptome analysis reveals the differentially expressed genes associated with growth in Guangxi partridge chickens. Genes 2022, 13, 798. [Google Scholar] [CrossRef]
- Yang, M.; Shi, L.; Ge, Y.; Leng, D.; Zeng, B.; Wang, T.; Jie, H.; Li, D. Dynamic changes in the gut microbial community and function during broiler growth. Microbiol. Spectr. 2022, 10, e01005-22. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-X.; Chen, L.; Ma, T.; Li, X.; Zheng, M.; Zhou, X.; Chen, L.; Qian, X.; Xi, J.; Lu, H.; et al. EasyAmplicon: An easy-to-use, open-source, reproducible, and community-based pipeline for amplicon data analysis in microbiome research. iMeta 2023, 2, e83. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Li, D.Y.; Zhang, L.; Smith, D.G.; Xu, H.L.; Liu, Y.P.; Zhao, X.L.; Wang, Y.; Zhu, Q. Genetic effects of melatonin receptor genes on chicken reproductive traits. Czech J. Anim. Sci. 2013, 58, 58–64. [Google Scholar] [CrossRef]
- Huang, S.; Sheng, P.; Zhang, H. Isolation and identification of cellulolytic bacteria from the gut of Holotrichia parallela larvae (coleoptera: Scarabaeidae). Int. J. Mol. Sci. 2012, 13, 2563–2577. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Deng, J.; Yang, Z.; Zeng, L.; Yang, X. Metagenomic analysis reveals linkages between cecal microbiota and feed efficiency in Xiayan chickens. Poult. Sci. 2020, 99, 7066–7075. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Chen, Y.-T.; Hsieh, H.-H.; Chen, M.-J. Effect of Lactobacillus mali APS1 and L. kefiranofaciens M1 on obesity and glucose homeostasis in diet-induced obese mice. J. Funct. Foods 2016, 23, 580–589. [Google Scholar] [CrossRef]
- Souillard, R.; Laurentie, J.; Kempf, I.; Le Caër, V.; Le Bouquin, S.; Serror, P.; Allain, V. Increasing incidence of Enterococcus-associated diseases in poultry in France over the past 15 years. Vet. Microbiol. 2022, 269, 109426. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Q.; Wu, X.; Qi, H.; Das, R.; Lin, H.; Shi, J.; Wang, S.; Yang, J.; Xue, Y.; et al. Vancomycin exposure caused opportunistic pathogens bloom in intestinal microbiome by simulator of the human intestinal microbial ecosystem (SHIME). Environ. Pollut. 2020, 265, 114399. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.V.; Torres, N.; Tovar, A.R. PPAR-α as a key nutritional and environmental sensor for metabolic adaptation. Adv. Nutr. 2013, 4, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Moll, F.; Walter, M.; Rezende, F.; Helfinger, V.; Vasconez, E.; De Oliveira, T.; Greten, F.R.; Olesch, C.; Weigert, A.; Radeke, H.H.; et al. NoxO1 controls proliferation of colon epithelial cells. Front. Immunol. 2018, 9, 973. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, G.; Qu, G.; Liu, B.; Zhang, X.; Li, G.; Jin, N.; Li, C.; Bai, J.; Zhao, C. Effects of dietary Lactobacillus rhamnosus GG supplementation on the production performance, egg quality, eggshell ultrastructure, and lipid metabolism of late-phase laying hens. BMC Vet. Res. 2023, 19, 150. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Zhou, Z.; Yu, L.; Jiang, K.; Xia, J.; Mi, Y.; Zhang, C.; Li, J. Lactobacillus salivarius and Lactobacillus agilis feeding regulates intestinal stem cells activity by modulating crypt niche in hens. Appl. Microbiol. Biotechnol. 2021, 105, 8823–8835. [Google Scholar] [CrossRef] [PubMed]
- Gorreja, F.; Rush, S.T.; Kasper, D.L.; Meng, D.; Walker, W.A. The developmentally regulated fetal enterocyte gene, ZP4, mediates anti-inflammation by the symbiotic bacterial surface factor polysaccharide A on Bacteroides fragilis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G398–G407. [Google Scholar] [CrossRef]
- Fatahi-Bafghi, M. Characterization of the Rothia Spp. and their role in human clinical infections. Infect. Genet. Evol. 2021, 93, 104877. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.M.; Gage, D.J. Development and application of aerobic, chemically defined media for Dysgonomonas. Anaerobe 2021, 67, 102302. [Google Scholar] [CrossRef]
- Cai, J.; Sun, L.; Gonzalez, F.J. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe 2022, 30, 289–300. [Google Scholar] [CrossRef]
- Jeong, Y.-J.; Park, H.-Y.; Nam, H.-K.; Lee, K.-W. Fermented Maillard reaction products by Lactobacillus Gasseri 4M13 alters the intestinal microbiota and improves dysfunction in type 2 diabetic mice with colitis. Pharmaceuticals 2021, 14, 299. [Google Scholar] [CrossRef]
- Zhang, J.; Yi, C.; Han, J.; Ming, T.; Zhou, J.; Lu, C.; Li, Y.; Su, X. Novel High-docosahexaenoic-acid tuna oil supplementation modulates gut microbiota and alleviates obesity in high-fat diet mice. Food Sci. Nutr. 2020, 8, 6513–6527. [Google Scholar] [CrossRef]
- Kakiuchi, N.; Yoshida, K.; Uchino, M.; Kihara, T.; Akaki, K.; Inoue, Y.; Kawada, K.; Nagayama, S.; Yokoyama, A.; Yamamoto, S.; et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 2020, 577, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Fu, Q.; Zhou, S.; Song, L.; Ren, Y.; Dong, X.; Su, B.; Li, C. The Mucosal expression signatures of G-type lysozyme in turbot (Scophthalmus maximus) following bacterial challenge. Fish Shellfish Immunol. 2016, 54, 612–619. [Google Scholar] [CrossRef]
- Shan, Y.; Fang, C.; Cheng, C.; Wang, Y.; Peng, J.; Fang, W. Immersion infection of germ-free Zebrafish with listeria monocytogenes induces transient expression of innate immune response genes. Front. Microbiol. 2015, 6, 373. [Google Scholar] [CrossRef] [PubMed]
- Hinton, A.; Hume, M.E.; Deloach, J.R. Role of metabolic intermediates in the inhibition of Salmonella typhimurium and Salmonella enteritidis by Veillonella. J. Food Prot. 1993, 56, 932–937. [Google Scholar] [CrossRef]
- Song, Y.; Cui, Y.; Wang, Y.; Yu, J.; Wang, B.; Wen, Q.; Zheng, X. Donor selection for fecal bacterial transplantation and its combined effects with inulin on early growth and ileal development in chicks. J. Appl. Microbiol. 2023, 134, lxad099. [Google Scholar] [CrossRef] [PubMed]
- Zeebone, Y.Y.; Bóta, B.; Halas, V.; Libisch, B.; Olasz, F.; Papp, P.; Keresztény, T.; Gerőcs, A.; Ali, O.; Kovács, M.; et al. Gut-faecal microbial and health-marker response to dietary fumonisins in weaned Pigs. Toxins 2023, 15, 328. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Wang, H.; Li, M.; Rong, J.; Liao, X.; Wu, Y.; Wang, Y. Differences in peripheral and central metabolites and gut microbiome of laying hens with different feather-pecking phenotypes. Front. Microbiol. 2023, 14, 1132866. [Google Scholar] [CrossRef]
- Song, B.; Li, P.; Yan, S.; Liu, Y.; Gao, M.; Lv, H.; Lv, Z.; Guo, Y. Effects of dietary astragalus polysaccharide supplementation on the Th17/Treg balance and the gut microbiota of broiler chickens challenged with necrotic enteritis. Front. Immunol. 2022, 13, 781934. [Google Scholar] [CrossRef]
- Quinteros, J.A.; Scott, P.C.; Wilson, T.B.; Anwar, A.M.; Scott, T.; Muralidharan, C.; Van, T.T.H.; Moore, R.J. Isoquinoline alkaloids induce partial protection of laying hens from the impact of Campylobacter hepaticus (spotty liver disease) challenge. Poult. Sci. 2021, 100, 101423. [Google Scholar] [CrossRef]
- Yokoyama, M.; Hinode, D.; Yoshioka, M.; Fukui, M.; Tanabe, S.; Grenier, D.; Ito, H.-O. Relationship between Campylobacter rectus and periodontal status during pregnancy. Oral Microbiol. Immunol. 2008, 23, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Watanabe-Yanai, A.; Tamamura-Andoh, Y.; Arai, N.; Akiba, M.; Kusumoto, M. Tryptanthrin reduces Campylobacter jejuni colonization in the chicken gut by a bactericidal mechanism. Appl. Environ. Microbiol. 2023, 89, e0170122. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.B.; Melvin, W.J.; Audu, C.O.; Bame, M.; Rhoads, N.; Wu, W.; Kanthi, Y.; Knight, J.S.; Adili, R.; Holinstat, M.A.; et al. The histone methyltransferase MLL1/KMT2A in monocytes drives coronavirus-associated coagulopathy and inflammation. Blood 2023, 141, 725–742. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Seballos, S.; Fletcher, B.; Romigh, T.; Yehia, L.; Mester, J.; Senter, L.; Niazi, F.; Saji, M.; Ringel, M.D.; et al. Germline compound heterozygous poly-glutamine deletion in USF3 may be involved in predisposition to heritable and sporadic epithelial thyroid carcinoma. Hum. Mol. Genet. 2017, 26, 243–257. [Google Scholar] [CrossRef]
- Smith, J.; Speed, D.; Hocking, P.M.; Talbot, R.T.; Degen, W.G.; Schijns, V.E.; Glass, E.J.; Burt, D.W. Development of a chicken 5 K microarray targeted towards immune function. BMC Genom. 2006, 7, 49. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, K.; Liu, X.; Duan, Y.; Jiang, X.; Li, N.; Du, Y.; Li, D.; Feng, C. Dynamic Changes in Intestinal Gene Expression and Microbiota across Chicken Egg-Laying Stages. Animals 2024, 14, 1529. https://doi.org/10.3390/ani14111529
Shi K, Liu X, Duan Y, Jiang X, Li N, Du Y, Li D, Feng C. Dynamic Changes in Intestinal Gene Expression and Microbiota across Chicken Egg-Laying Stages. Animals. 2024; 14(11):1529. https://doi.org/10.3390/ani14111529
Chicago/Turabian StyleShi, Kai, Xiangping Liu, Ying Duan, Xusheng Jiang, Ni Li, Yuesong Du, Dongfeng Li, and Chungang Feng. 2024. "Dynamic Changes in Intestinal Gene Expression and Microbiota across Chicken Egg-Laying Stages" Animals 14, no. 11: 1529. https://doi.org/10.3390/ani14111529
APA StyleShi, K., Liu, X., Duan, Y., Jiang, X., Li, N., Du, Y., Li, D., & Feng, C. (2024). Dynamic Changes in Intestinal Gene Expression and Microbiota across Chicken Egg-Laying Stages. Animals, 14(11), 1529. https://doi.org/10.3390/ani14111529