
Complete Mitochondrial Genomes of Nannostomus Pencilfish: Genome Characterization and Phylogenetic Analysis

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitogenome Sequencing and Assembly
2.3. Sequence Analysis
2.4. Phylogenetic Analysis
3. Results
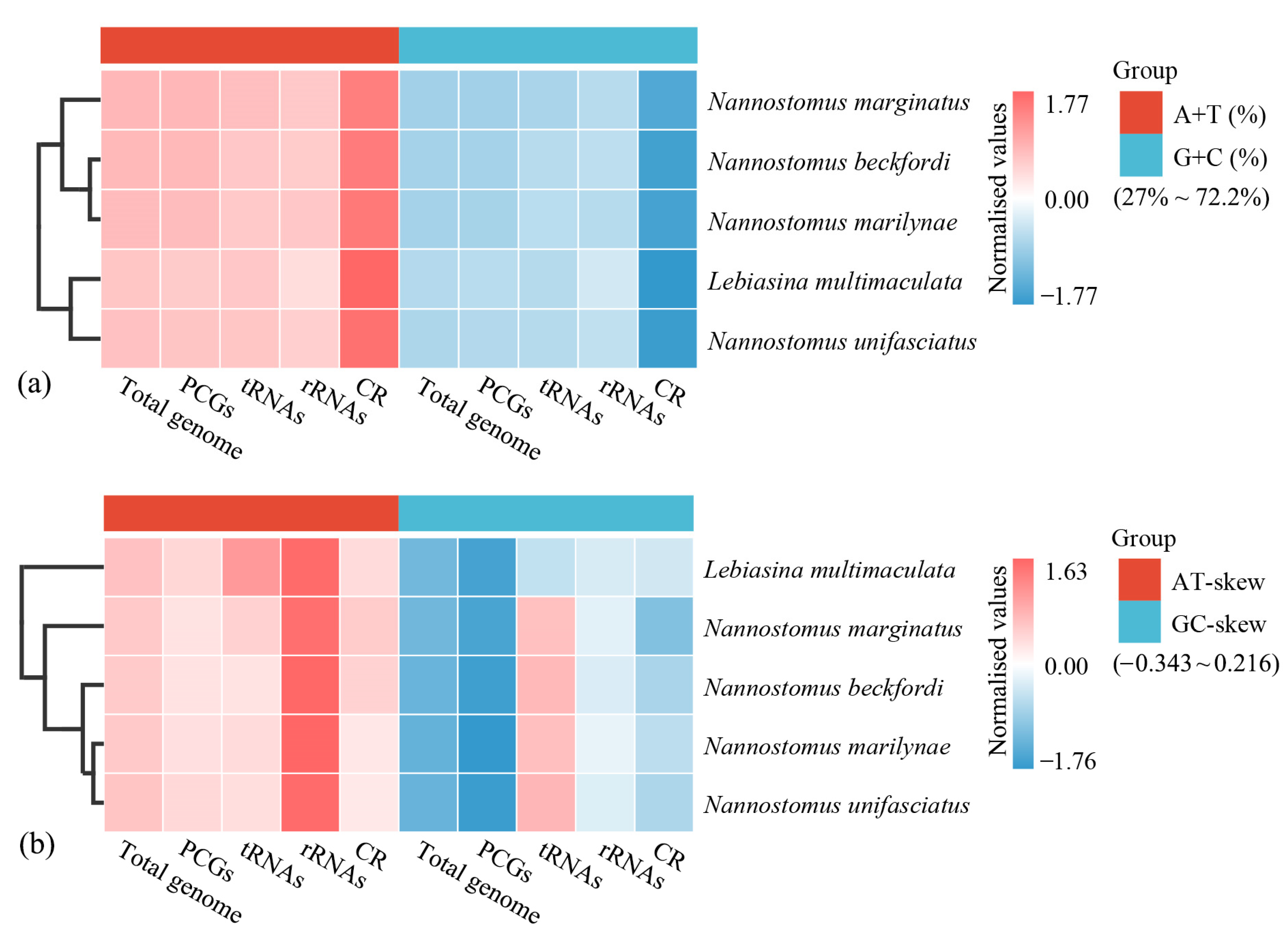
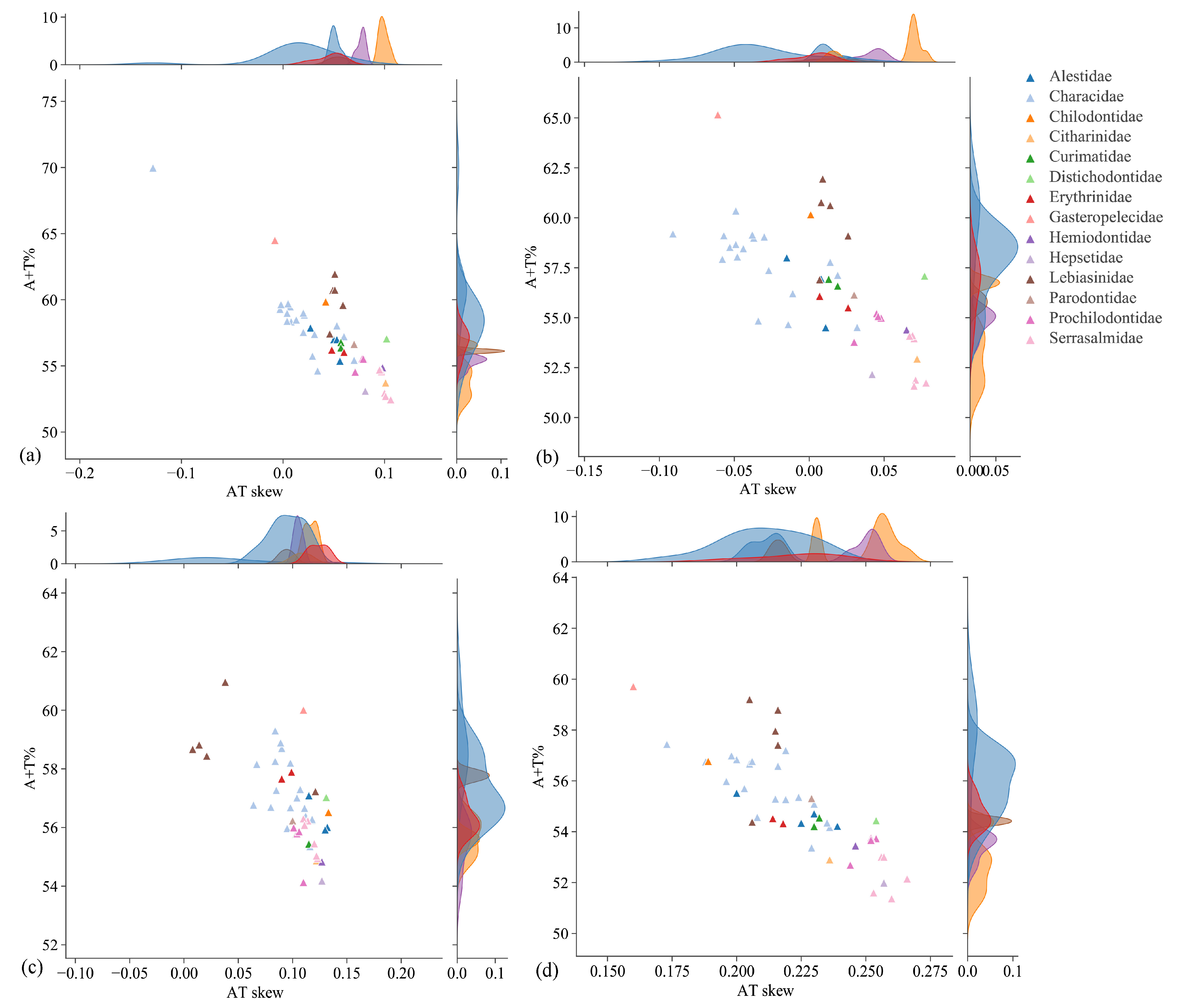
3.1. Genome Organization and Composition
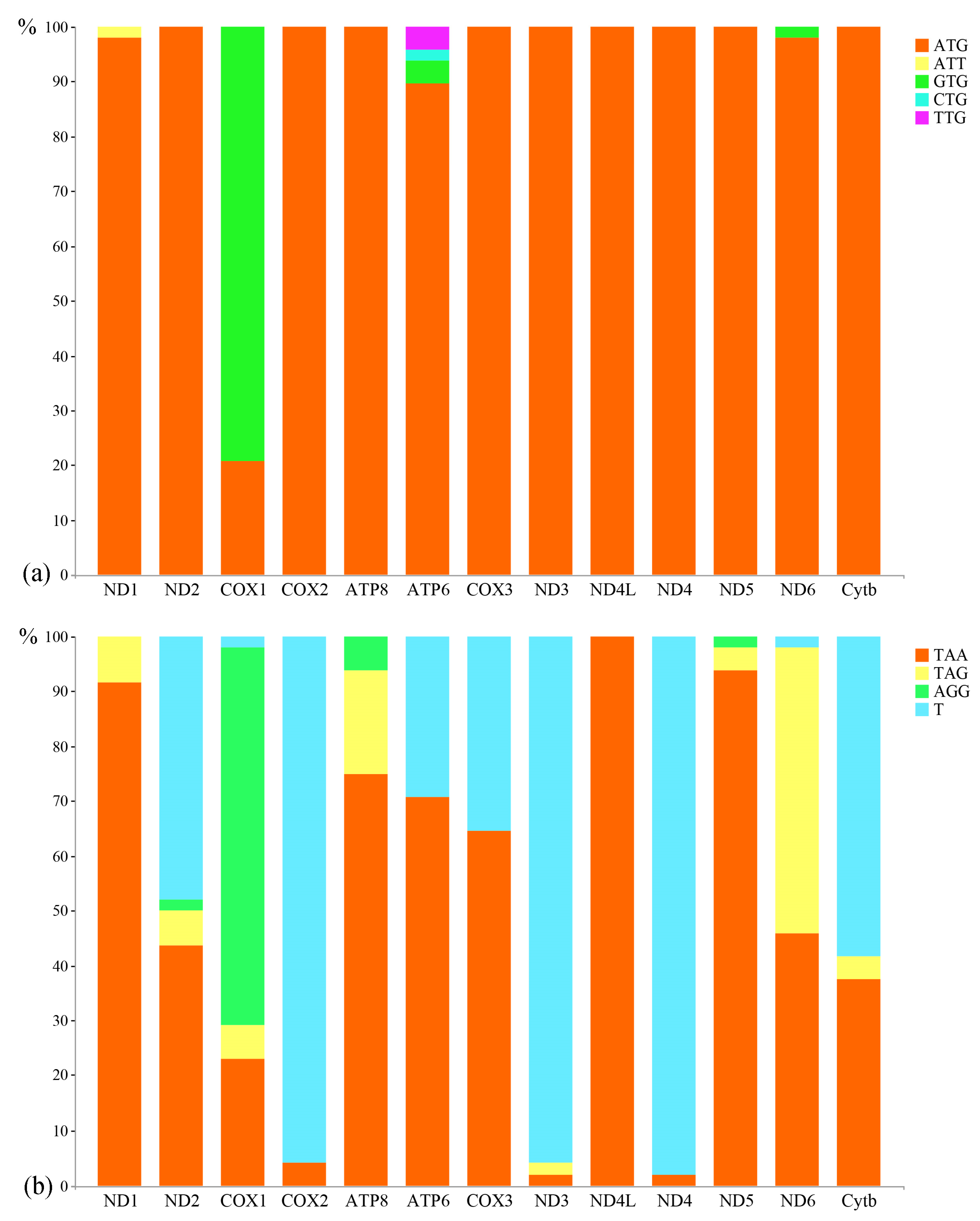
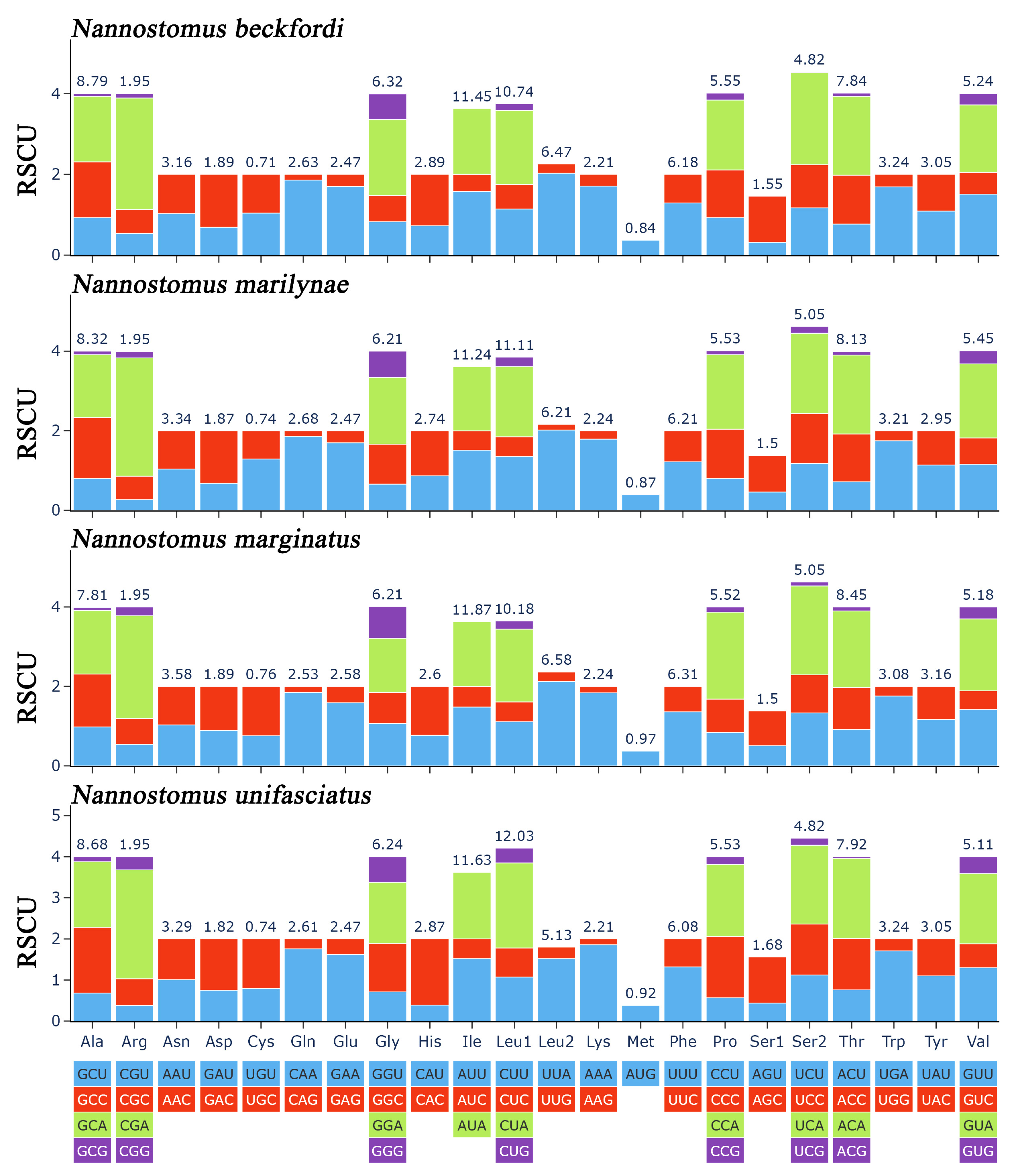
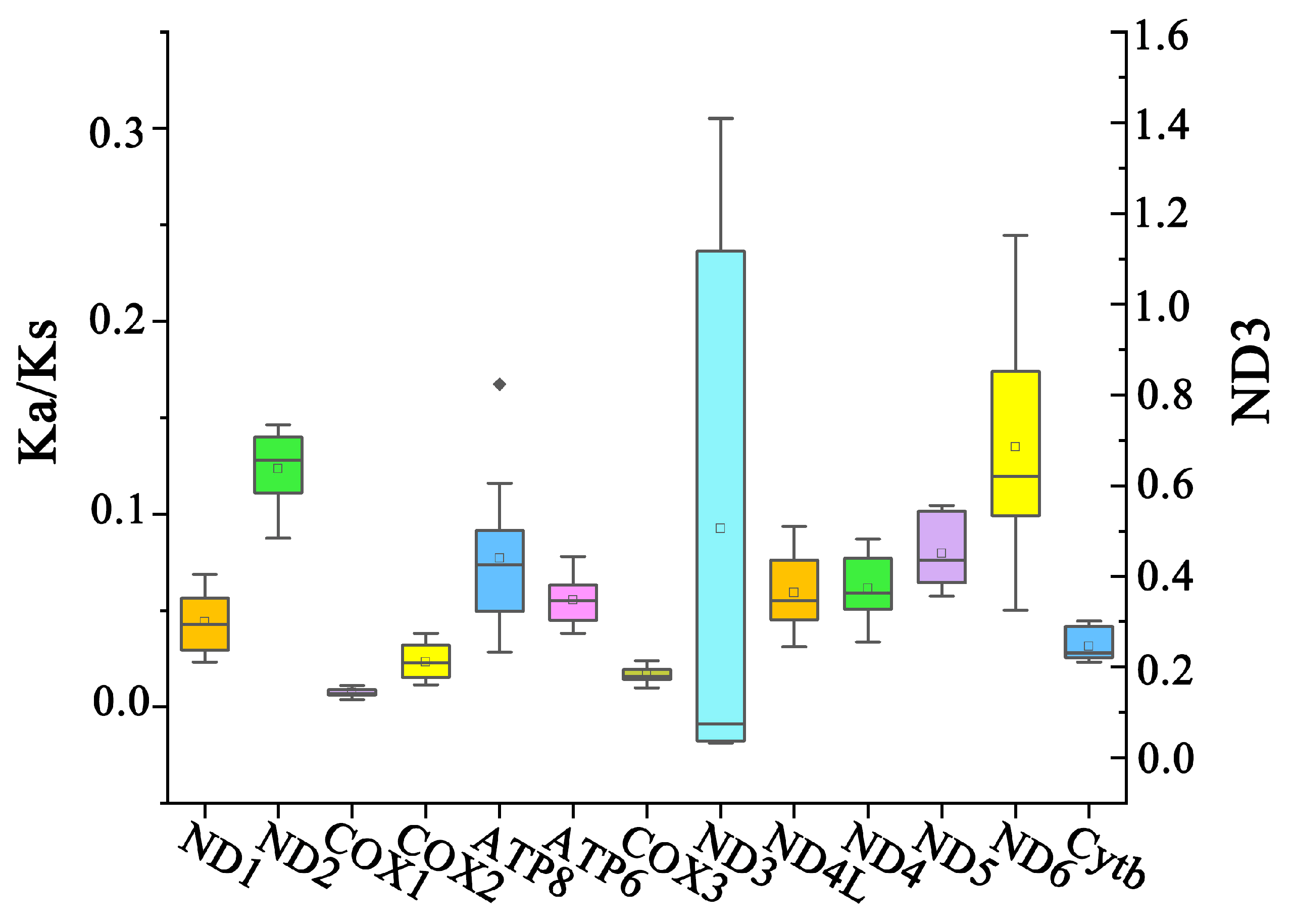
3.2. Protein-Coding Genes and Codon Usage
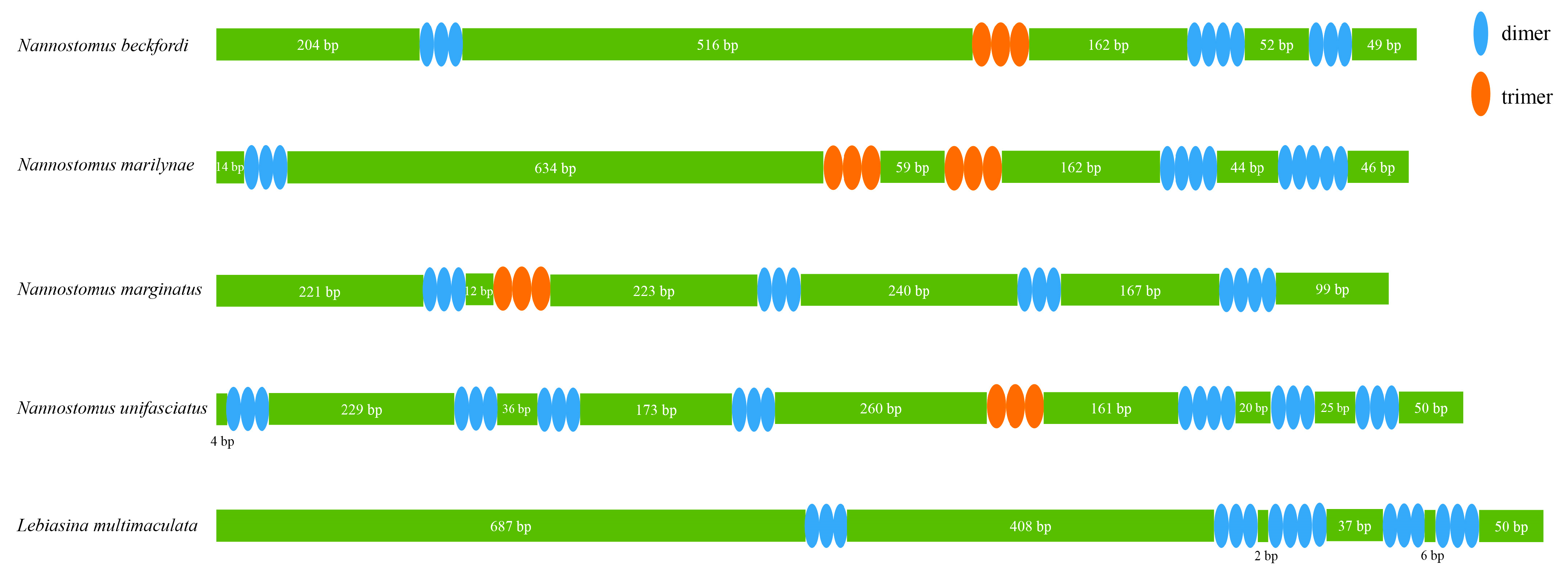
3.3. rRNA, tRNA Genes, and CR
3.4. Phylogenetic Relationships
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benzaquem, D.C.; Oliveira, C.; Batista, J.d.S.; Zuanon, J.; Porto, J.I.R. DNA Barcoding in Pencilfishes (Lebiasinidae: Nannostomus) Reveals Cryptic Diversity across the Brazilian Amazon. PLoS ONE 2015, 10, e0112217. [Google Scholar] [CrossRef] [PubMed]
- Sember, A.; de Oliveira, E.A.; Ráb, P.; Bertollo, L.A.C.; de Freitas, N.L.; Viana, P.F.; Yano, C.F.; Hatanaka, T.; Marinho, M.M.F.; de Moraes, R.L.R.; et al. Centric Fusions behind the Karyotype Evolution of Neotropical Nannostomus Pencilfishes (Characiforme, Lebiasinidae): First Insights from a Molecular Cytogenetic Perspective. Genes 2020, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Beheregaray, L.B.; Schwartz, T.S.; Möller, L.M.; Call, D.; Chao, N.L.; Caccone, A. A set of microsatellite DNA markers for the one-lined pencilfish Nannostomus unifasciatus, an Amazonian flooded forest fish. Mol. Ecol. Notes 2004, 4, 333–335. [Google Scholar] [CrossRef]
- Terencio, M.L.; Schneider, C.H.; Porto, J.I.R. Molecular signature of the D-loop in the brown pencilfish Nannostomus eques (Characiformes, Lebiasinidae) reveals at least two evolutionary units in the Rio Negro basin, Brazil. J. Fish Biol. 2012, 81, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, A.L.B.; Daga, V.S.; Bezerra, L.A.V.; Vitule, J.R.S.; Jacobi, C.M.; Silva, L.G.M. All the colors of the world: Biotic homogenization-differentiation dynamics of freshwater fish communities on demand of the Brazilian aquarium trade. Hydrobiologia 2020, 847, 3897–3915. [Google Scholar] [CrossRef]
- Meng, F.; Huang, Y.; Liu, B.; Zhu, K.; Zhang, J.; Jing, F.; Xia, L.; Liu, Y. The complete mitochondrial genome of Lebiasina astrigata (Characiformes: Lebiasinida) and phylogenetic studies of Characiformes. Mitochondrial DNA Part B 2019, 4, 579–580. [Google Scholar] [CrossRef]
- Sassi, F.d.M.C.; Hatanaka, T.; de Moraes, R.L.R.; Toma, G.A.; de Oliveira, E.A.; Liehr, T.; Rab, P.; Bertollo, L.A.C.; Viana, P.F.; Feldberg, E.; et al. An Insight into the Chromosomal Evolution of Lebiasinidae (Teleostei, Characiformes). Genes 2020, 11, 365. [Google Scholar] [CrossRef]
- Calcagnotto, D.; Schaefer, S.A.; DeSalle, R. Relationships among characiform fishes inferred from analysis of nuclear and mitochondrial gene sequences. Mol. Phylogenetics Evol. 2005, 36, 135–153. [Google Scholar] [CrossRef] [PubMed]
- de Pinna, M.; Zuanon, J.; Py-Daniel, L.R.; Petry, P. A new family of neotropical freshwater fishes from deep fossorial Amazonian habitat, with a reappraisal of morphological characiform phylogeny (Teleostei: Ostariophysi). Zool. J. Linn. Soc. 2018, 182, 76–106. [Google Scholar] [CrossRef]
- Xu, W.; Lin, S.; Liu, H. Mitochondrial genomes of five Hyphessobrycon tetras and their phylogenetic implications. Ecol. Evol. 2021, 11, 12754–12764. [Google Scholar] [CrossRef]
- Wang, J.; Xu, W.; Liu, Y.; Bai, Y.; Liu, H. Comparative mitochondrial genomics and phylogenetics for species of the snakehead genus Channa Scopoli, 1777 (Perciformes: Channidae). Gene 2023, 857, 147186. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Xu, W.; Wang, J.; Xu, R.; Jiang, H.; Ding, J.; Wu, H.; Wu, Y.; Liu, H. Comparative mitochondrial genomics of tetras: Insights into phylogenetic relationships in Characidae. Biologia 2022, 77, 2905–2914. [Google Scholar] [CrossRef]
- Chang, Y.-S.; Huang, F.-L.; Lo, T.-B. The complete nucleotide sequence and gene organization of carp (Cyprinus carpio) mitochondrial genome. J. Mol. Evol. 1994, 38, 138–155. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-F.; Wang, Z.-Y.; Wang, Z.-L.; Yu, X.-P. Complete mitochodrial genome of the crab spider Ebrechtella tricuspidata (Araneae: Thomisidae): A novel tRNA rearrangement and phylogenetic implications for Araneae. Genomics 2019, 111, 1266–1273. [Google Scholar] [CrossRef]
- Zhu, S.-R.; Fu, J.-J.; Wang, Q.; Li, J.-L. Identification of Channa species using the partial cytochrome c oxidase subunit I (COI) gene as a DNA barcoding marker. Biochem. Syst. Ecol. 2013, 51, 117–122. [Google Scholar] [CrossRef]
- Serrao, N.R.; Steinke, D.; Hanner, R.H. Calibrating Snakehead Diversity with DNA Barcodes: Expanding Taxonomic Coverage to Enable Identification of Potential and Established Invasive Species. PLoS ONE 2014, 9, e99546. [Google Scholar] [CrossRef]
- Powell, A.F.; Barker, F.K.; Lanyon, S.M. Empirical evaluation of partitioning schemes for phylogenetic analyses of mitogenomic data: An avian case study. Mol. Phylogenet. Evol. 2013, 66, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Chandhini, S.; Vargheese, S.; Philip, S.; Kumar, V.J.R. Deciphering the mitochondrial genome of Malabar snakehead, Channa diplogramma (Teleostei; Channidae). Biologia 2020, 75, 741–748. [Google Scholar] [CrossRef]
- Hu, Y.; Xing, W.; Hu, Z.; Liu, G. Phylogenetic Analysis and Substitution Rate Estimation of Colonial Volvocine Algae Based on Mitochondrial Genomes. Genes 2020, 11, 115. [Google Scholar] [CrossRef]
- Casal, C.M.V. Global Documentation of Fish Introductions: The Growing Crisis and Recommendations for Action. Biol. Invasions 2006, 8, 3–11. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Diao, C.; Xi, Y.; Xiao, T. Identification and analysis of key genes in osteosarcoma using bioinformatics. Oncol. Lett. 2018, 15, 2789–2794. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Ranwez, V.; Douzery, E.J.P.; Cambon, C.; Chantret, N.; Delsuc, F. MACSE v2: Toolkit for the Alignment of Coding Sequences Accounting for Frameshifts and Stop Codons. Mol. Biol. Evol. 2018, 35, 2582–2584. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Liddle, A.R. Information criteria for astrophysical model selection. Mon. Not. R. Astron. Soc. Lett. 2007, 377, L74–L78. [Google Scholar] [CrossRef]
- Yang, Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Xu, K.; Zhao, F.; Liu, W.; Li, L.; Hua, Z.; Zhou, X. itol.toolkit accelerates working with iTOL (Interactive Tree of Life) by an automated generation of annotation files. Bioinformatics 2023, 39, btad339. [Google Scholar] [CrossRef] [PubMed]
- Montaña-Lozano, P.; Moreno-Carmona, M.; Ochoa-Capera, M.; Medina, N.S.; Boore, J.L.; Prada, C.F. Comparative genomic analysis of vertebrate mitochondrial reveals a differential of rearrangements rate between taxonomic class. Sci. Rep. 2022, 12, 5479. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Qiu, G.; Cao, C.; Wang, X.; Jiang, L.; Zhang, T.; Geng, Z.; Jin, S. Mitochondrial genome and phylogenetic analysis of Chaohu duck. Gene 2023, 851, 147018. [Google Scholar] [CrossRef] [PubMed]
- Pasa, R.; Menegídio, F.B.; Rodrigues-Oliveira, I.H.; da Silva, I.B.; de Campos, M.L.C.B.; Rocha-Reis, D.A.; Heslop-Harrison, J.S.; Schwarzacher, T.; Kavalco, K.F. Ten Complete Mitochondrial Genomes of Gymnocharacini (Stethaprioninae, Characiformes). Insights Into Evolutionary Relationships and a Repetitive Element in the Control Region (D-loop). Front. Ecol. Evol. 2021, 9, 650783. [Google Scholar] [CrossRef]
- Zhu, Q.; Luo, S.; Pan, S.; Su, X.; Liu, Z.; Chen, J. Complete mitogenome of Gymnocorymbus ternetzi (Characiformes: Characidae: Gymnocorymbus) and phylogenetic implications. Mitochondrial DNA Part B 2022, 7, 58–59. [Google Scholar] [CrossRef]
- Liu, H.; Sun, C.; Zhu, Y.; Li, Y.; Wei, Y.; Ruan, H. Mitochondrial genomes of four American characins and phylogenetic relationships within the family Characidae (Teleostei: Characiformes). Gene 2020, 762, 145041. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tai, J.; Zhang, W.; He, K.; Lan, H.; Liu, H. Comparison of seven complete mitochondrial genomes from Lamprologus and Neolamprologus (Chordata, Teleostei, Perciformes) and the phylogenetic implications for Cichlidae. ZooKeys 2023, 1184, 115–132. [Google Scholar] [CrossRef]
- Silva, D.M.Z.d.A.; Utsunomia, R.; Ruiz-Ruano, F.J.; Oliveira, C.; Foresti, F. The complete mitochondrial genome sequence of Astyanax paranae (Teleostei: Characiformes). Mitochondrial DNA Part B 2016, 1, 586–587. [Google Scholar] [CrossRef]
- Kundu, S.; Kang, H.-E.; Kim, A.R.; Lee, S.R.; Kim, E.-B.; Amin, M.H.F.; Andriyono, S.; Kim, H.-W.; Kang, K. Mitogenomic Characterization and Phylogenetic Placement of African Hind, Cephalopholis taeniops: Shedding Light on the Evolution of Groupers (Serranidae: Epinephelinae). Int. J. Mol. Sci. 2024, 25, 1822. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Yang, K.; Ye, L.; Gao, X. Complete Mitogenome and Phylogenetic Analyses of Galerita orientalis Schmidt-Goebel, 1846 (Insecta: Coleoptera: Carabidae: Galeritini). Genes 2022, 13, 2199. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yi, H.; Zhu, Y. Novel insights into adaptive evolution based on the unusual AT-skew in Acheilognathus gracilis mitogenome and phylogenetic relationships of bitterling. Gene 2024, 902, 148154. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Yang, Z.; Liu, C.; Lee, X.; Zhu, K. Characterization of the Complete Mitochondrial Genome of the Spotted Catfish Arius maculatus (Thunberg, 1792) and Its Phylogenetic Implications. Genes 2022, 13, 2128. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Hassanin, A.; Léger, N.; Deutsch, J. Evidence for Multiple Reversals of Asymmetric Mutational Constraints during the Evolution of the Mitochondrial Genome of Metazoa, and Consequences for Phylogenetic Inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ding, J.; Lin, S.; Xu, R.; Liu, H. Comparative mitogenomes of three species in Moenkhausia: Rare irregular gene rearrangement within Characidae. Int. J. Biol. Macromol. 2021, 183, 1079–1086. [Google Scholar] [CrossRef]
- Padhi, A. Geographic variation within a tandemly repeated mitochondrial DNA D-loop region of a North American freshwater fish, Pylodictis olivaris. Gene 2014, 538, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Mirande, J.M. Morphology, molecules and the phylogeny of Characidae (Teleostei, Characiformes). Cladistics 2019, 35, 282–300. [Google Scholar] [CrossRef]
- Lima, F.C.; Albrecht, M.P.; Pavanelli, C.S.; Vono, V. Threatened fishes of the world: Brycon nattereri Günther, 1864 (Characidae). Environ. Biol. Fishes 2008, 83, 207–208. [Google Scholar] [CrossRef]
- de Siqueira-Silva, D.H.; Silva, A.P.d.S.; Costa, R.d.S.; Senhorini, J.A.; Ninhaus-Silveira, A.; Veríssimo-Silveira, R. Preliminary study on testicular germ cell isolation and transplantation in an endangered endemic species Brycon orbignyanus (Characiformes: Characidae). Fish Physiol. Biochem. 2021, 47, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Travenzoli, N.M.; Silva, P.C.; Santos, U.; Zanuncio, J.C.; Oliveira, C.; Dergam, J.A. Cytogenetic and Molecular Data Demonstrate that the Bryconinae (Ostariophysi, Bryconidae) Species from Southeastern Brazil Form a Phylogenetic and Phylogeographic Unit. PLoS ONE 2015, 10, e0137843. [Google Scholar] [CrossRef] [PubMed]
- Arruda, P.S.S.; Ferreira, D.C.; Oliveira, C.; Venere, P.C. DNA Barcoding Reveals High Levels of Divergence among Mitochondrial Lineages of Brycon (Characiformes, Bryconidae). Genes 2019, 10, 639. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Avelino, G.S.; Abe, K.T.; Mariguela, T.C.; Benine, R.C.; Ortí, G.; Vari, R.P.; e Castro, R.M.C. Phylogenetic relationships within the speciose family Characidae (Teleostei: Ostariophysi: Characiformes) based on multilocus analysis and extensive ingroup sampling. BMC Evol. Biol. 2011, 11, 275. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.; Cortinas, M.N.; Musto, H. The Analysis of Protein Coding Genes Suggests Monophyly ofTrypanosoma. Mol. Phylogenet. Evol. 1996, 5, 333–343. [Google Scholar] [CrossRef]
- Han, J.-H.; Cho, M.-H.; Kim, S.B. Ribosomal and protein coding gene based multigene phylogeny on the family Streptomycetaceae. Syst. Appl. Microbiol. 2012, 35, 1–6. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order. | Family | Genus | Species | Size (bp) | Accession No. |
|---|---|---|---|---|---|
| Characiformes | Alestidae | Abramites | Abramites hypselonotus | 16,685 | MW541938.1 |
| Megaleporinus | Megaleporinus elongatus | 16,774 | KU980144.1 | ||
| Megaleporinus obtusidens | 16,682 | KY825191.1 | |||
| Phenacogrammus | Phenacogrammus interruptus | 16,652 | AB054129.1 | ||
| Bryconidae | Brycon | Brycon henni | 16,885 | KP027535.1 | |
| Brycon nattereri | 16,837 | MT428073.1 | |||
| Brycon orbignyanus | 16,802 | KY825192.1 | |||
| Salminus | Salminus brasiliensis | 17,721 | KM245047.1 | ||
| Characidae | Astyanax | Astyanax aeneus | 16,769 | BK013055.1 | |
| Astyanax mexicanus | 16,768 | BK013062.1 | |||
| Hemigrammus | Hemigrammus armstrongi | 16,789 | MW742324.1 | ||
| Hemigrammus bleheri | 17,021 | LC074360.1 | |||
| Hemigrammus erythrozonus | 16,710 | MT484070.1 | |||
| Hyphessobrycon | Hyphessobrycon amapaensis | 17,824 | MW742322.1 | ||
| Hyphessobrycon elachys | 17,224 | MW315747.1 | |||
| Hyphessobrycon flammeus | 16,008 | MW315748.1 | |||
| Hyphessobrycon herbertaxelrodi | 17,417 | MT769327.1 | |||
| Hyphessobrycon pulchripinnis | 17,618 | MW331227.1 | |||
| Moenkhausia | Moenkhausia costae | 15,811 | MW366831.1 | ||
| Moenkhausia sanctaefilomenae | 18,437 | MW407181.1 | |||
| Paracheirodon | Paracheirodon axelrodi | 17,100 | AB898197.1 | ||
| Paracheirodon innesi | 16,962 | KT783482.1 | |||
| Chilodontidae | Chilodus | Chilodus punctatus | 16,869 | AP011984.1 | |
| Citharinidae | Citharinus | Citharinus congicus | 16,453 | AP011985.1 | |
| Curimatidae | Curimata | Curimata mivartii | 16,705 | KP025764.1 | |
| Curimatopsis | Curimatopsis evelynae | 16,779 | AP011988.1 | ||
| Distichodontidae | Distichodus | Distichodus sexfasciatus | 16,555 | AB070242.1 | |
| Erythrinidae | Hoplias | Hoplias intermedius | 16,629 | KU523584.1 | |
| Hoplias malabaricus | 16,638 | AP011992.1 | |||
| Gasteropelecidae | Carnegiella | Carnegiella strigata | 17,852 | AP011983.1 | |
| Hemiodontidae | Hemiodopsis | Hemiodopsis gracilis | 16,731 | AP011990.1 | |
| Hepsetidae | Hepsetus | Hepsetus odoe | 16,803 | AP011991.1 | |
| Lebiasinidae | Lebiasina | Lebiasina multimaculata | 16,899 | AP006766.1 | |
| Nannostomus | Nannostomus beckfordi | 16,690 | OR857846 | ||
| Nannostomus marilynae | 16,667 | OR857847 | |||
| Nannostomus marginatus | 16,661 | OR857848 | |||
| Nannostomus unifasciatus | 16,681 | OR857849 | |||
| Parodontidae | Apareiodon | Apareiodon affinis | 16,679 | AP011998.1 | |
| Prochilodontidae | Ichthyoelephas | Ichthyoelephas longirostris | 16,840 | KP025763.1 | |
| Prochilodus | Prochilodus argenteus | 16,697 | KR014816.1 | ||
| Prochilodus costatus | 16,699 | KR014817.1 | |||
| Prochilodus lineatus | 16,699 | KM245045.1 | |||
| Serrasalmidae | Colossoma | Colossoma macropomum | 16,703 | KP188830.1 | |
| Metynnis | Metynnis hypsauchen | 16,737 | MH358334.1 | ||
| Myloplus | Myloplus rubripinnis | 16,662 | MH358336.1 | ||
| Piaractus | Piaractus brachypomus | 16,722 | KJ993871.2 | ||
| Piaractus mesopotamicus | 16,722 | KM245046.1 | |||
| Pygocentrus | Pygocentrus nattereri | 16,706 | AP012000.1 | ||
| Cypriniformes | Cyprinidae | Cyprinus | Cyprinus carpio | 16,592 | OL693871.1 |
| Gene | Position | Size (bp) | Orientation | Codon | Intergenic Nucleotides (bp) | ||
|---|---|---|---|---|---|---|---|
| From | To | Initiation | Termination | ||||
| tRNA-Phe | 1/1/1/1 | 70/70/71/70 | 70/70/7/70 | +/+/+/+ | 0/0/0/0 | ||
| 12S rRNA | 71/71/72/71 | 1026/1025/1024/1024 | 956/955/953/954 | +/+/+/+ | 0/0/0/0 | ||
| tRNA-Val | 1026/1025/1024/1024 | 1097/1096/1095/1095 | 72/72/72/72 | +/+/+/+ | −1/−1/−1/−1 | ||
| 16S rRNA | 1098/1097/1096/1096 | 2793/2786/2777/2785 | 1696/1690/1682/1690 | +/+/+/+ | 0/0/0/0 | ||
| tRNA-Leu | 2796/2789/2779/2788 | 2871/2864/2854/2863 | 76/76/76/76 | +/+/+/+ | 2/2/1/2 | ||
| ND1 | 2872/2865/2852/2864 | 3846/3839/3826/3838 | 975/975/975/975 | +/+/+/+ | ATG/ATG/ATT/ATG | TAG/TAA/TAA/TAA | 0/0/−3/0 |
| tRNA-Ile | 3851/3843/3832/3842 | 3922/3914/3903/3913 | 72/72/72/72 | +/+/+/+ | 4/3/5/3 | ||
| tRNA-Gln | 3921/3913/3902/3912 | 3991/3983/3972/3982 | 71/71/71/71 | −/−/−/− | −2/−2/−2/−2 | ||
| tRNA-Met | 3992/3983/3973/3986 | 4061/4052/4042/4055 | 70/70/70/70 | +/+/+/+ | 0/−1/0/3 | ||
| ND2 | 4062/4053/4043/4056 | 5106/5099/5087/5100 | 1045/1047/1045/1045 | +/+/+/+ | ATG/ATG/ATG/ATG | T/TAG/T/T | 0/0/0/0 |
| tRNA-Trp | 5107/5098/5088/5101 | 5178/5169/5160/5172 | 72/72/73/72 | +/+/+/+ | 0/−2/0/0 | ||
| tRNA-Ala | 5182/5173/5163/5176 | 5250/5241/5231/5244 | 69/69/69/69 | −/−/−/− | 3/3/2/3 | ||
| tRNA-Asn | 5252/5243/5233/5246 | 5324/5315/5305/5318 | 73/73/73/73 | −/−/−/− | 1/1/1/1 | ||
| tRNA-Cys | 5354/5346/5337/5349 | 5420/5411/5402/5414 | 67/66/66/66 | −/−/−/− | 29/30/31/30 | ||
| tRNA-Tyr | 5421/5412/5403/5415 | 5490/5481/5470/5483 | 70/70/68/69 | −/−/−/− | 0/0/0/0 | ||
| COX1 | 5492/5483/5472/5485 | 7048/7039/7028/7041 | 1557/1557/1557/1557 | +/+/+/+ | GTG/GTG/GTG/GTG | AGG/AGG/AGG/AGG | 1/1/1/1 |
| tRNA-Ser | 7040/7031/7020/7033 | 7110/7101/7090/7103 | 71/71/71/71 | −/−/−/− | −9/−9/−9/−9 | ||
| tRNA-Asp | 7115/7106/7096/7108 | 7184/7175/7165/7177 | 70/70/70/70 | +/+/+/+ | 4/4/5/4 | ||
| COX2 | 7198/7189/7180/7192 | 7888/7879/7870/7882 | 691/691/691/691 | +/+/+/+ | ATG/ATG/ATG/ATG | T/T/T/T | 13/13/14/14 |
| tRNA-Lys | 7889/7880/7871/7883 | 7962/7953/7944/7956 | 74/74/74/74 | +/+/+/+ | 0/0/0/0 | ||
| ATP8 | 7964/7955/7946/7958 | 8131/8122/8113/8125 | 168/168/168/168 | +/+/+/+ | ATG/ATG/ATG/ATG | TAA/TAA/TAA/TAA | 1/1/1/1 |
| ATP6 | 8122/8113/8104/8116 | 8805/8796/8787/8799 | 684/684/684/684 | +/+/+/+ | ATG/ATG/ATG/ATG | TAA/TAA/TAA/TAA | −10/−10/−10/−10 |
| COX3 | 8805/8796/8787/8799 | 9590/9581/9572/9584 | 786/786/786/786 | +/+/+/+ | ATG/ATG/ATG/ATG | TAA/TAA/TAA/TAA | −1/−1/−1/−1 |
| tRNA-Gly | 9590/9581/9572/9584 | 9662/9652/9647/9655 | 73/72/76/72 | +/+/+/+ | −1/−1/−1/−1 | ||
| ND3 | 9663/9653/9648/9656 | 10,008/9998/9993/10,001 | 346/346/346/346 | +/+/+/+ | ATG/ATG/ATG/ATG | T/T/T/T | 0/0/0/0 |
| tRNA-Arg | 10,009/9999/9994/10,002 | 10,079/10,067/10,063/10,071 | 71/69/70/70 | +/+/+/+ | 0/0/0/0 | ||
| ND4L | 10,080/10,068/10,064/10,072 | 10,376/10,364/10,360/10,368 | 297/297/297/297 | +/+/+/+ | ATG/ATG/ATG/ATG | TAA/TAA/TAA/TAA | 0/0/0/0 |
| ND4 | 10,370/10,358/10,354/10,362 | 11,750/11,738/11,734/11,742 | 1381/1381/1381/1381 | +/+/+/+ | ATG/ATG/ATG/ATG | T/T/T/T | −7/−7/−7/−7 |
| tRNA-His | 11,752/11,740/11,735/11,744 | 11,821/11,808/11,804/11,813 | 70/69/70/70 | +/+/+/+ | 1/1/0/1 | ||
| tRNA-Ser | 11,822/11,809/11,805/11,814 | 11,889/11,876/11,872/11,881 | 68/68/68/68 | +/+/+/+ | 0/0/0/0 | ||
| tRNA-Leu | 11,891/11,878/11,874/11,883 | 11,963/11,950/11,946/11,955 | 73/73/73/73 | +/+/+/+ | 1/1/1/1 | ||
| ND5 | 11,964/11,951/11,947/11,956 | 13,802/13,789/13,785/13,794 | 1839/1839/1839/1839 | +/+/+/+ | ATG/ATG/ATG/ATG | TAA/TAA/TAA/TAA | 0/0/0/0 |
| ND6 | 13,799/13,786/13,782/13,791 | 14,317/14,304/14,300/14,309 | 519/519/519/519 | −/−/−/− | ATG/ATG/ATG/ATG | TAG/TAA/TAG/TAA | −4/−4/−4/−4 |
| tRNA-Glu | 14,318/14,305/14,301/14,310 | 14,386/14,372/14,369/14,377 | 69/68/69/68 | −/−/−/− | 0/0/0/0 | ||
| Cytb | 14,392/14,378/14,375/14,383 | 15,534/15,520/15,518/15,525 | 1143/1143/1144/1143 | +/+/+/+ | ATG/ATG/ATG/ATG | TAA/TAA/T/TAA | 5/5/5/5 |
| tRNA-Thr | 15,536/15,522/15,519/15,527 | 15,607/15,595/15,592/15,599 | 72/74/74/73 | +/+/+/+ | 1/1/0/1 | ||
| tRNA-Pro | 15,609/15,597/15,596/15,601 | 15,678/15,666/15,664/15,670 | 70/70/69/70 | −/−/−/− | 1/1/3/1 | ||
| CR | 15,679/15,667/15,665/15,671 | 16,690/16,667/16,661/16,681 | 1012/1001/997/1011 | 0/0/0/0 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, W.; Tai, J.; He, K.; Xu, T.; Zhang, G.; Xu, B.; Liu, H. Complete Mitochondrial Genomes of Nannostomus Pencilfish: Genome Characterization and Phylogenetic Analysis. Animals 2024, 14, 1598. https://doi.org/10.3390/ani14111598
Xu W, Tai J, He K, Xu T, Zhang G, Xu B, Liu H. Complete Mitochondrial Genomes of Nannostomus Pencilfish: Genome Characterization and Phylogenetic Analysis. Animals. 2024; 14(11):1598. https://doi.org/10.3390/ani14111598
Chicago/Turabian StyleXu, Wei, Jingzhe Tai, Ke He, Tangjun Xu, Gaoji Zhang, Boyu Xu, and Hongyi Liu. 2024. "Complete Mitochondrial Genomes of Nannostomus Pencilfish: Genome Characterization and Phylogenetic Analysis" Animals 14, no. 11: 1598. https://doi.org/10.3390/ani14111598
APA StyleXu, W., Tai, J., He, K., Xu, T., Zhang, G., Xu, B., & Liu, H. (2024). Complete Mitochondrial Genomes of Nannostomus Pencilfish: Genome Characterization and Phylogenetic Analysis. Animals, 14(11), 1598. https://doi.org/10.3390/ani14111598