
Study on Changes in Gut Microbiota and Microbiability in Rabbits at Different Developmental Stages

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal and Experimental Design
2.3. DNA Extraction and Sequencing
2.4. Bioinformatics Analysis
2.5. Statistical Analysis
3. Results
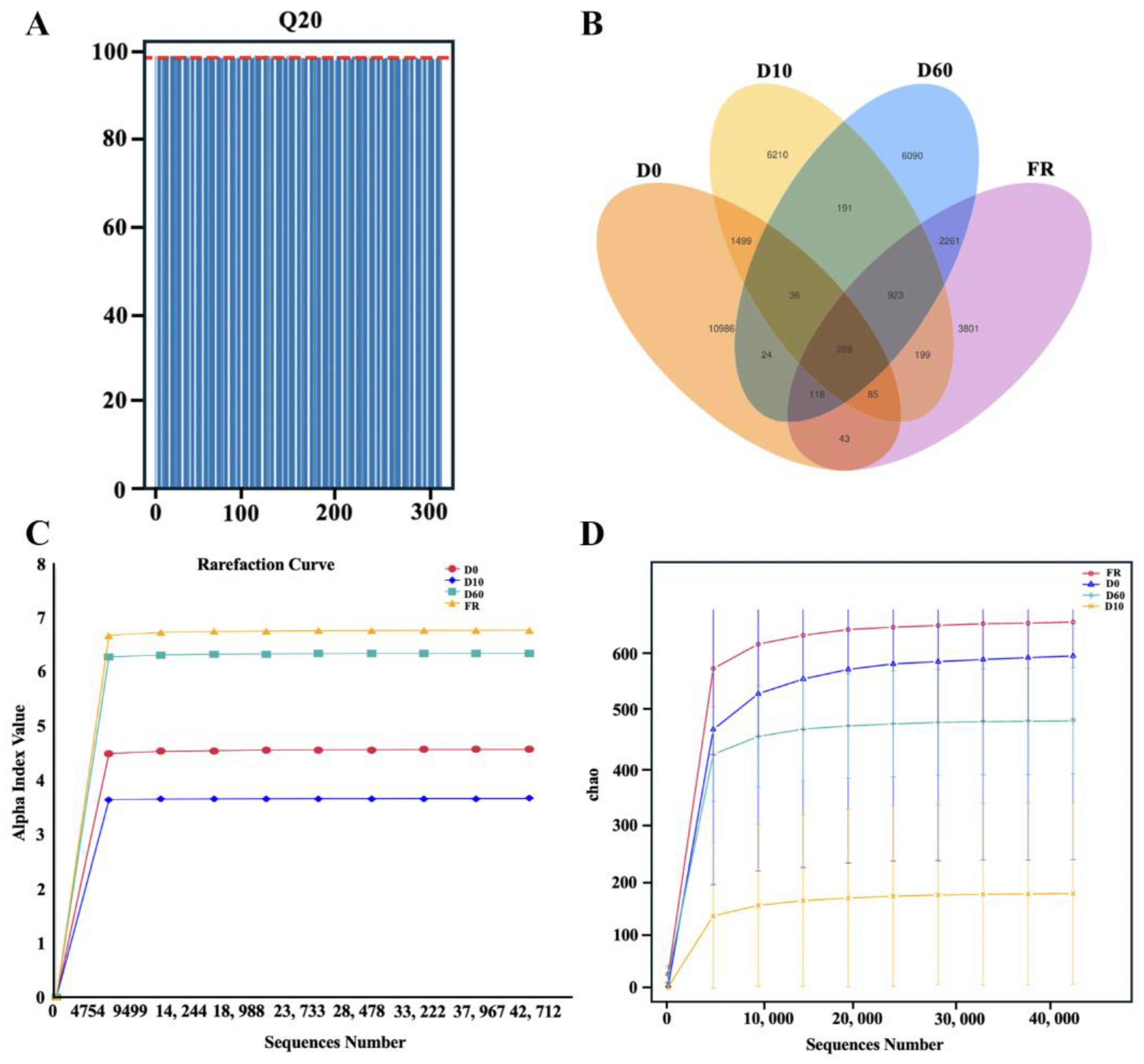
3.1. Data Evaluation
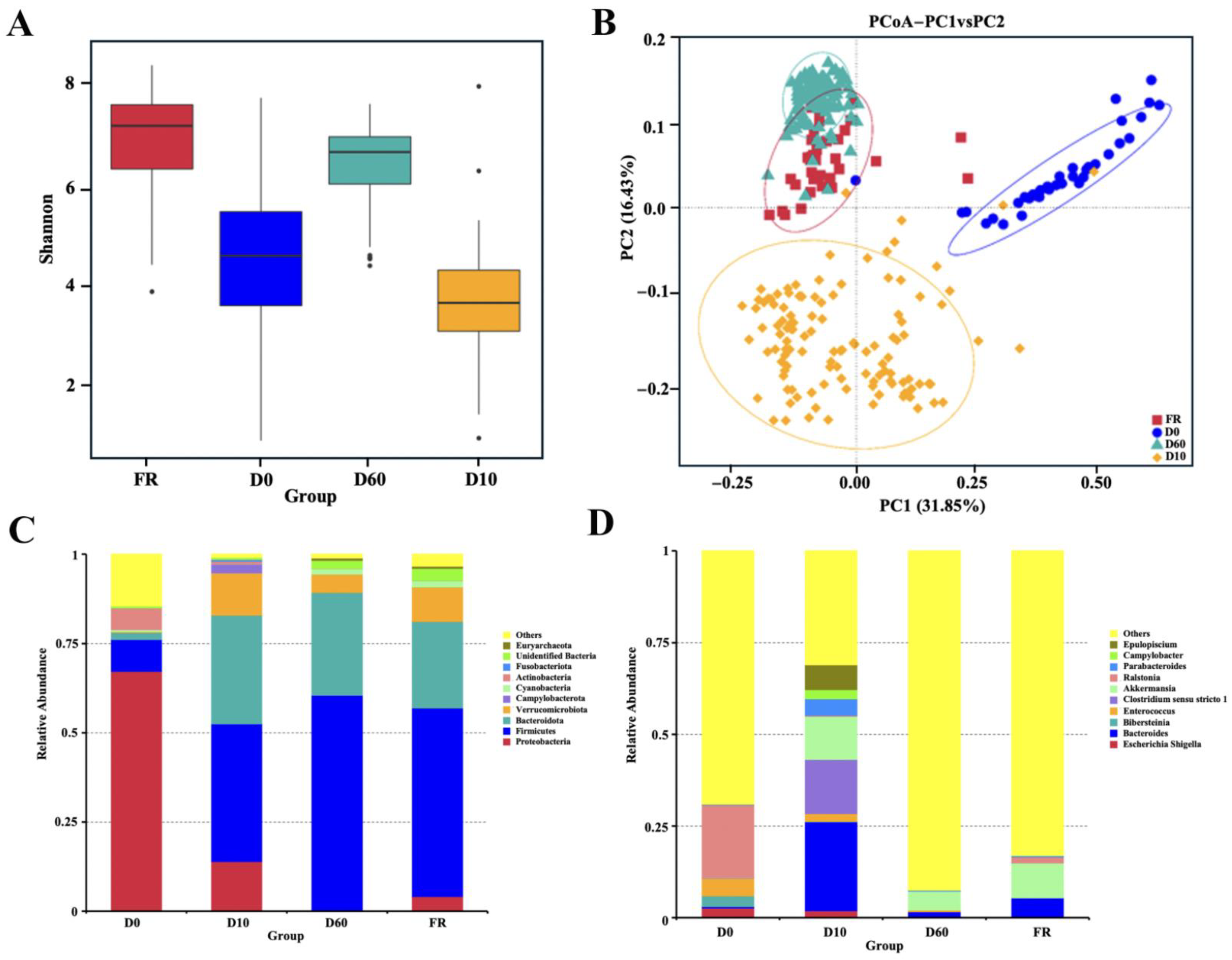
3.2. Composition and Comparison of Gut Microbiota in Female Rabbits and Their Different Aged Juvenile Rabbits
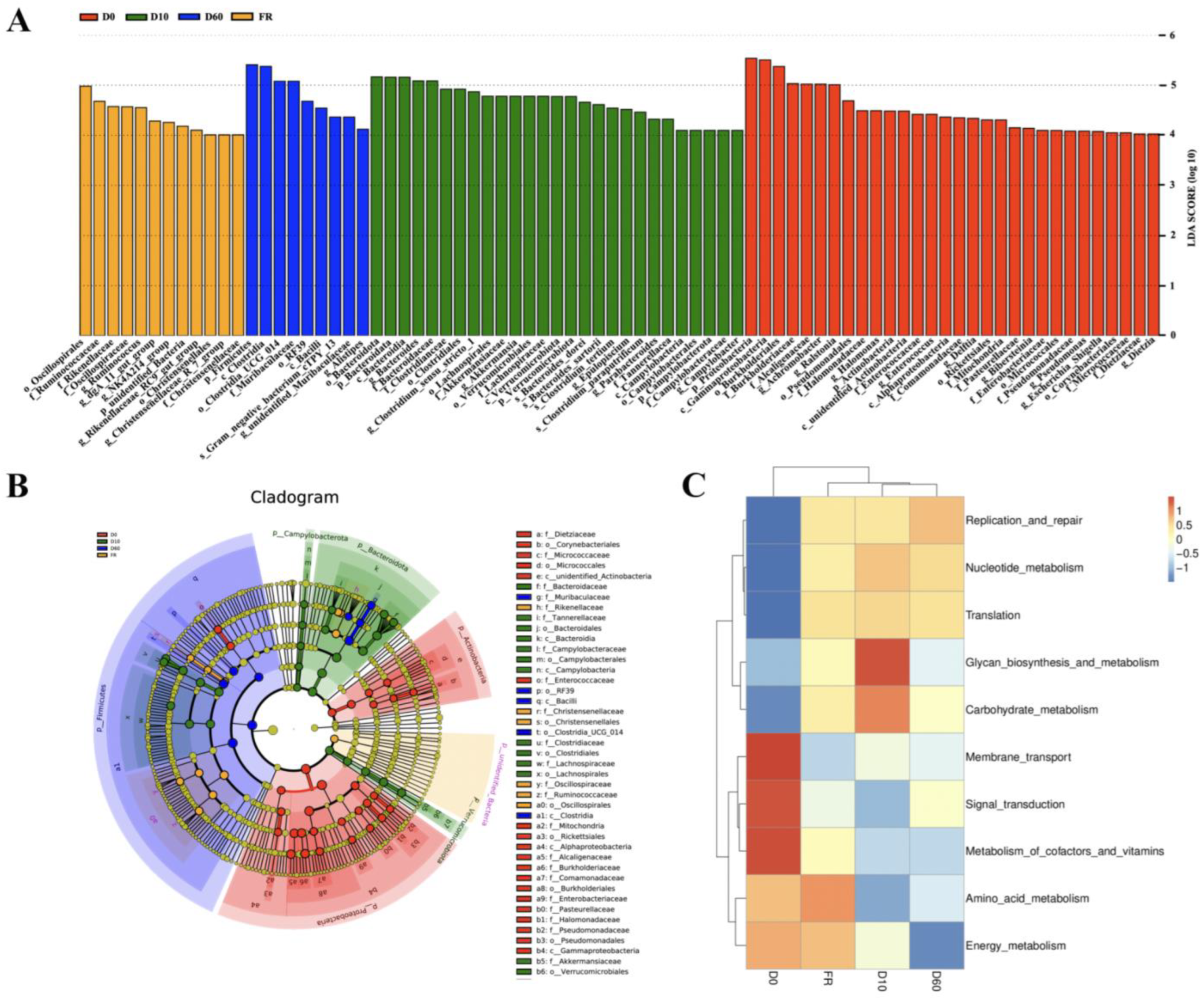
3.3. Identification and Functional Prediction of Biomarkers for Female Rabbits and Their Different Aged Juvenile Rabbits
3.4. The Influence of Gut Microbiota in Female Rabbits on the Establishment of Gut Microbiota in Offspring
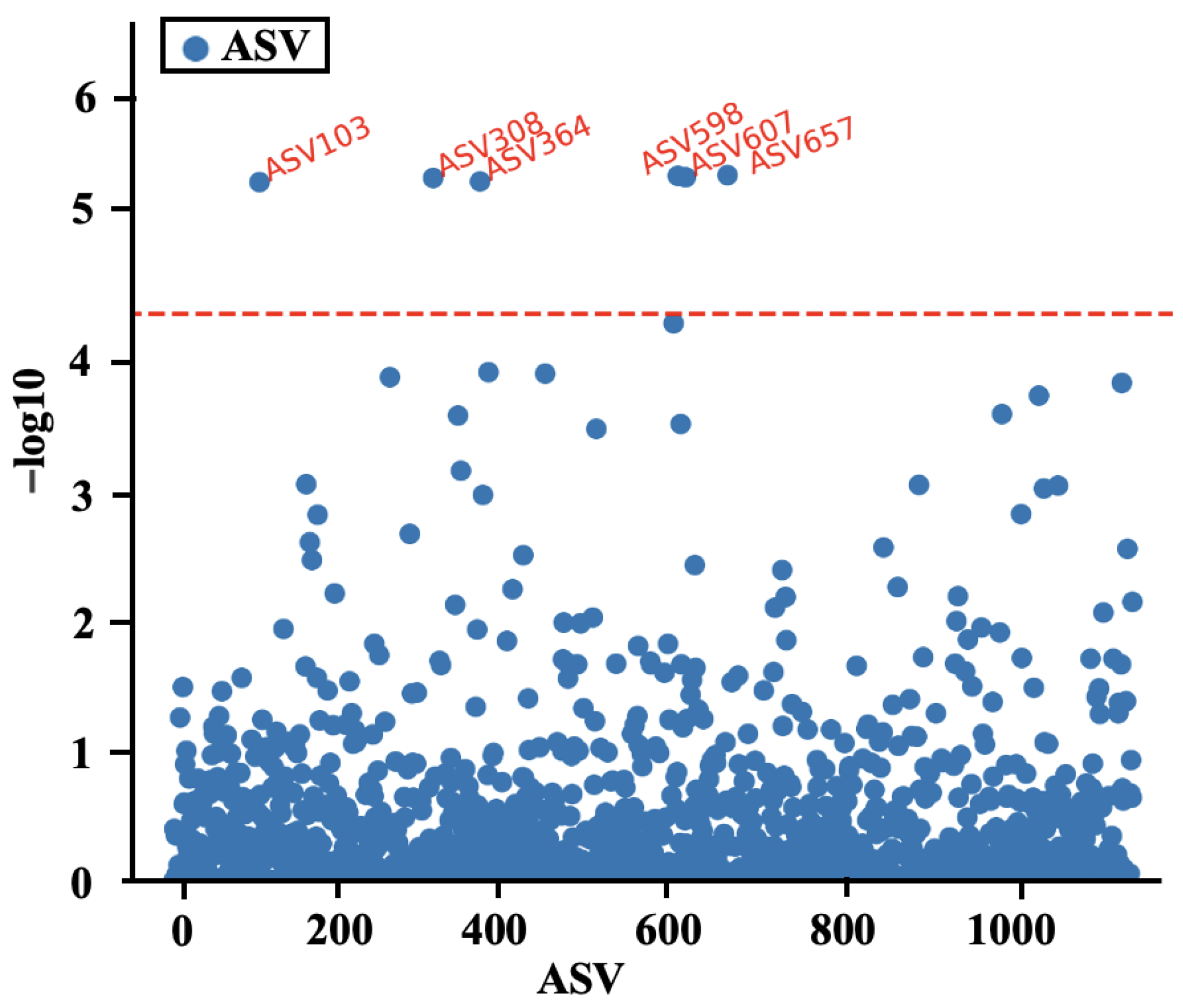
3.5. Correlation Analysis between the Relative Abundance of Gut Microbiota and Body Weight in 60-Day-Old Rabbits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Makki, K.; Deehan, E.C.; Walter, J.; Bäckhed, F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.P. Impact of animal health programmes on poverty reduction and sustainable livestock development. Rev. Sci. Tech. 2017, 36, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kong, Q.; Li, X.; Zhao, J.; Zhang, H.; Chen, W.; Wang, G. A High-Fat Diet Increases Gut Microbiota Biodiversity and Energy Expenditure Due to Nutrient Difference. Nutrients 2020, 12, 3197. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the gut microbiome and mucosal immune system. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Rao, M.C.; Chang, E.B. Gut microbiota as a transducer of dietary cues to regulate host circadian rhythms and metabolism. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Hase, K. Gut microbiota-generated metabolites in animal health and disease. Nat. Chem. Biol. 2014, 10, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Cui, Y.; Ali, Q.; Zhu, X.; Li, D.; Ma, S.; Wang, Z.; Wang, C.; Shi, Y. Gut Microbiota Modulate Rabbit Meat Quality in Response to Dietary Fiber. Front. Nutr. 2022, 9, 849429. [Google Scholar] [CrossRef] [PubMed]
- Mancini, S.; Paci, G. Probiotics in Rabbit Farming: Growth Performance, Health Status, and Meat Quality. Animals 2021, 11, 3388. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhao, F.; Tian, H.; Li, J.; Guo, D. Effects of the dietary digestible fiber-to-starch ratio on pellet quality, growth and cecal microbiota of Angora rabbits. Asian Australas. J. Anim. Sci. 2020, 33, 623–633. [Google Scholar] [CrossRef]
- Fang, S.; Chen, X.; Pan, J.; Chen, Q.; Zhou, L.; Wang, C.; Xiao, T.; Gan, Q.F. Dynamic distribution of gut microbiota in meat rabbits at different growth stages and relationship with average daily gain (ADG). BMC Microbiol. 2020, 20, 116. [Google Scholar] [CrossRef]
- Yassour, M.; Jason, E.; Hogstrom, L.J.; Arthur, T.D.; Tripathi, S.; Siljander, H.; Selvenius, J.; Oikarinen, S.; Hyöty, H.; Virtanen, S.M.; et al. Strain-Level Analysis of Mother-to-Child Bacterial Transmission during the First Few Months of Life. Cell Host Microbe 2018, 24, 146–154.e144. [Google Scholar] [CrossRef] [PubMed]
- Reddivari, L.; Veeramachaneni, D.N.R.; Walters, W.A.; Lozupone, C.; Palmer, J.; Hewage, M.K.K.; Bhatnagar, R.; Amir, A.; Kennett, M.J.; Knight, R.; et al. Perinatal Bisphenol A Exposure Induces Chronic Inflammation in Rabbit Offspring via Modulation of Gut Bacteria and Their Metabolites. mSystems 2017, 2, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Dai, R.; Yang, L.; He, C.; Xu, K.; Liu, S.; Zhao, W.; Xiao, L.; Luo, L.; Zhang, Y.; et al. Inheritance and Establishment of Gut Microbiota in Chickens. Front. Microbiol. 2017, 8, 1967. [Google Scholar] [CrossRef] [PubMed]
- Savietto, D.; Paës, C.; Cauquil, L.; Fortun-Lamothe, L.; Combes, S. Evolution of gut microbial community through reproductive life in female rabbits and investigation of the link with offspring survival. Animal 2020, 14, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Trevelline, B.K.; Kohl, K.D. The gut microbiome influences host diet selection behavior. Proc. Natl. Acad. Sci. USA 2022, 119, e2117537119. [Google Scholar] [CrossRef] [PubMed]
- Mithieux, G. Gut Microbiota and Host Metabolism: What Relationship. Neuroendocrinology 2018, 106, 352–356. [Google Scholar] [CrossRef]
- Velasco-Galilea, M.; Piles, M.; Ramayo-Caldas, Y.; Varona, L.; Sánchez, J.P. Use of Bayes factors to evaluate the effects of host genetics, litter and cage on the rabbit cecal microbiota. Genet. Sel. Evol. 2022, 54, 46. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, D.; Legarra, A.; Tsuruta, S.; Masuda, Y.; Aguilar, I.; Misztal, I. Single-Step Genomic Evaluations from Theory to Practice: Using SNP Chips and Sequence Data in BLUPF90. Genes 2020, 11, 790. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liu, H.; Liu, M.; Yue, Z.; Li, C.; Liu, L.; Li, F. Metagenomics and metabolomics reveal that gut microbiome adapts to the diet transition in Hyla rabbits. Microbiol. Res. 2024, 283, 127705. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, C.; Li, F. Impact of dietary fiber/starch ratio in shaping caecal microbiota in rabbits. Can. J. Microbiol. 2015, 61, 771–784. [Google Scholar] [CrossRef]
- Monteils, V.; Cauquil, L.; Combes, S.; Godon, J.J.; Gidenne, T. Potential core species and satellite species in the bacterial community within the rabbit caecum. FEMS Microbiol. Ecol. 2008, 66, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Abecia, L.; Rodríguez-Romero, N.; Yañez-Ruiz, D.R.; Fondevila, M. Biodiversity and fermentative activity of caecal microbial communities in wild and farm rabbits from Spain. Anaerobe 2012, 18, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Cotozzolo, E.; Cremonesi, P.; Curone, G.; Menchetti, L.; Riva, F.; Biscarini, F.; Marongiu, M.L.; Castrica, M.; Castiglioni, B.; Miraglia, D.; et al. Characterization of Bacterial Microbiota Composition along the Gastrointestinal Tract in Rabbits. Animals 2020, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Guzman, C.E.; Bereza-Malcolm, L.T.; De Groef, B.; Franks, A.E. Presence of Selected Methanogens, Fibrolytic Bacteria, and Proteobacteria in the Gastrointestinal Tract of Neonatal Dairy Calves from Birth to 72 Hours. PLoS ONE 2015, 10, e0133048. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Kong, X.; Li, H.; Zhu, Q.; Guo, Q.; Yin, Y. Effects of dietary nutrient levels on microbial community composition and diversity in the ileal contents of pregnant Huanjiang mini-pigs. PLoS ONE 2017, 12, e0172086. [Google Scholar] [CrossRef]
- Shi, Y.; Tang, L.; Bai, X.; Du, K.; Wang, H.; Jia, X.; Lai, S. Heat Stress Altered the Vaginal Microbiome and Metabolome in Rabbits. Front. Microbiol. 2022, 13, 813622. [Google Scholar] [CrossRef]
- Hu, X.; Wang, F.; Yang, S.; Yuan, X.; Yang, T.; Zhou, Y.; Li, Y. Rabbit microbiota across the whole body revealed by 16S rRNA gene amplicon sequencing. BMC Microbiol. 2021, 21, 312. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Liu, J.; Lu, F.; Wang, L.; Li, Y.; Ouyang, C. Dynamic distribution of gallbladder microbiota in rabbit at different ages and health states. PLoS ONE 2019, 14, e0211828. [Google Scholar] [CrossRef]
- Beaumont, M.; Mussard, E.; Barilly, C.; Lencina, C.; Gress, L.; Painteaux, L.; Gabinaud, B.; Cauquil, L.; Aymard, P.; Canlet, C.; et al. Developmental Stage, Solid Food Introduction, and Suckling Cessation Differentially Influence the Comaturation of the Gut Microbiota and Intestinal Epithelium in Rabbits. J. Nutr. 2022, 152, 723–736. [Google Scholar] [CrossRef]
- Paës, C.; Gidenne, T.; Bébin, K.; Duperray, J.; Gohier, C.; Guené-Grand, E.; Rebours, G.; Barilly, C.; Gabinaud, B.; Cauquil, L.; et al. Early Introduction of Plant Polysaccharides Drives the Establishment of Rabbit Gut Bacterial Ecosystems and the Acquisition of Microbial Functions. mSystems 2022, 7, e0024322. [Google Scholar] [CrossRef]
- Vandenplas, Y.; Carnielli, V.P.; Ksiazyk, J.; Luna, M.S.; Migacheva, N.; Mosselmans, J.M.; Picaud, J.C.; Possner, M.; Singhal, A.; Wabitsch, M. Factors affecting early-life intestinal microbiota development. Nutrition 2020, 78, 110812. [Google Scholar] [CrossRef] [PubMed]
- Di Simone, N.; Santamaria Ortiz, A.; Specchia, M.; Tersigni, C.; Villa, P.; Gasbarrini, A.; Scambia, G.; D’Ippolito, S. Recent Insights on the Maternal Microbiota: Impact on Pregnancy Outcomes. Front. Immunol. 2020, 11, 528202. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, J.; Ren, E.; Su, Y.; Zhu, W. Co-occurrence of early gut colonization in neonatal piglets with microbiota in the maternal and surrounding delivery environments. Anaerobe 2018, 49, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Combes, S.; Gidenne, T.; Cauquil, L.; Bouchez, O.; Fortun-Lamothe, L. Coprophagous behavior of rabbit pups affects implantation of cecal microbiota and health status. J. Anim. Sci. 2014, 92, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Y.; Wang, K.; Li, Z.; Ji, M.; Li, F.; Han, L. Evaluating the effects of tannic acid on rabbit growth performance, digestibility, antioxidant status, intestinal morphology and caecal fermentation and microbiota. J. Anim. Physiol. Anim. Nutr. 2024, 108, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Piles, M.; Sánchez, J.P. Use of group records of feed intake to select for feed efficiency in rabbit. J. Anim. Breed. Genet. 2019, 136, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Chen, X.; Zhou, L.; Wang, C.; Chen, Q.; Lin, R.; Xiao, T.; Gan, Q. Faecal microbiota and functional capacity associated with weaning weight in meat rabbits. Microb. Biotechnol. 2019, 12, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.; Wang, J.; Xie, Y.; Zhang, J.; Guo, Y.; Tian, T.; Tang, J.; Ju, F.; Wang, C.; Liu, Y.; et al. Investigation of the growth performance, blood status, gut microbiome and metabolites of rabbit fed with low-nicotine tobacco. Front. Microbiol. 2022, 13, 1026680. [Google Scholar] [CrossRef]
- Guo, Z.; Ali, Q.; Abaidullah, M.; Gao, Z.; Diao, X.; Liu, B.; Wang, Z.; Zhu, X.; Cui, Y.; Li, D.; et al. High fat diet-induced hyperlipidemia and tissue steatosis in rabbits through modulating ileal microbiota. Appl. Microbiol. Biotechnol. 2022, 106, 7187–7207. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DIC | |||||||
|---|---|---|---|---|---|---|---|
| Model 1 | 840.99 ± 114.98 | 840.99 ± 114.98 | 1139.88 | ||||
| Model 2 | 176.07 ± 36.37 | 731.65 ± 148.54 | 907.72 ± 184.91 | 0.64 ± 0.17 | 0.19 ± 0.14 | 1082.73 | |
| Model 3 | 604.06 ± 217.91 | 338.12 ± 136.01 | 942.18 ± 152.93 | 1003.12 | |||
| Model 4 | 657.45 ± 204.01 | 146.59 ± 11.26 | 197.08 ± 126.37 | 1001.12 ± 240.24 | 0.66 ± 0.15 | 0.15 ± 0.12 | 969.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, C.; Ma, Y.; Xia, S.; Shao, J.; Tang, T.; Sun, W.; Jia, X.; Wang, J.; Lai, S. Study on Changes in Gut Microbiota and Microbiability in Rabbits at Different Developmental Stages. Animals 2024, 14, 1741. https://doi.org/10.3390/ani14121741
Fu C, Ma Y, Xia S, Shao J, Tang T, Sun W, Jia X, Wang J, Lai S. Study on Changes in Gut Microbiota and Microbiability in Rabbits at Different Developmental Stages. Animals. 2024; 14(12):1741. https://doi.org/10.3390/ani14121741
Chicago/Turabian StyleFu, Chong, Yue Ma, Siqi Xia, Jiahao Shao, Tao Tang, Wenqiang Sun, Xianbo Jia, Jie Wang, and Songjia Lai. 2024. "Study on Changes in Gut Microbiota and Microbiability in Rabbits at Different Developmental Stages" Animals 14, no. 12: 1741. https://doi.org/10.3390/ani14121741