
Effects of Freshwater Acidification on the Gut Microbial Community of Trachemys scripta elegans
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Research Animals
2.2. DNA Extraction and PCR Amplification
2.3. Processing of Sequencing Data
2.4. Ecological and Statistical Analysis
3. Results
3.1. Analysis of 16S rRNA Sequencing Results
3.2. Alpha and Beta Diversity Analyses
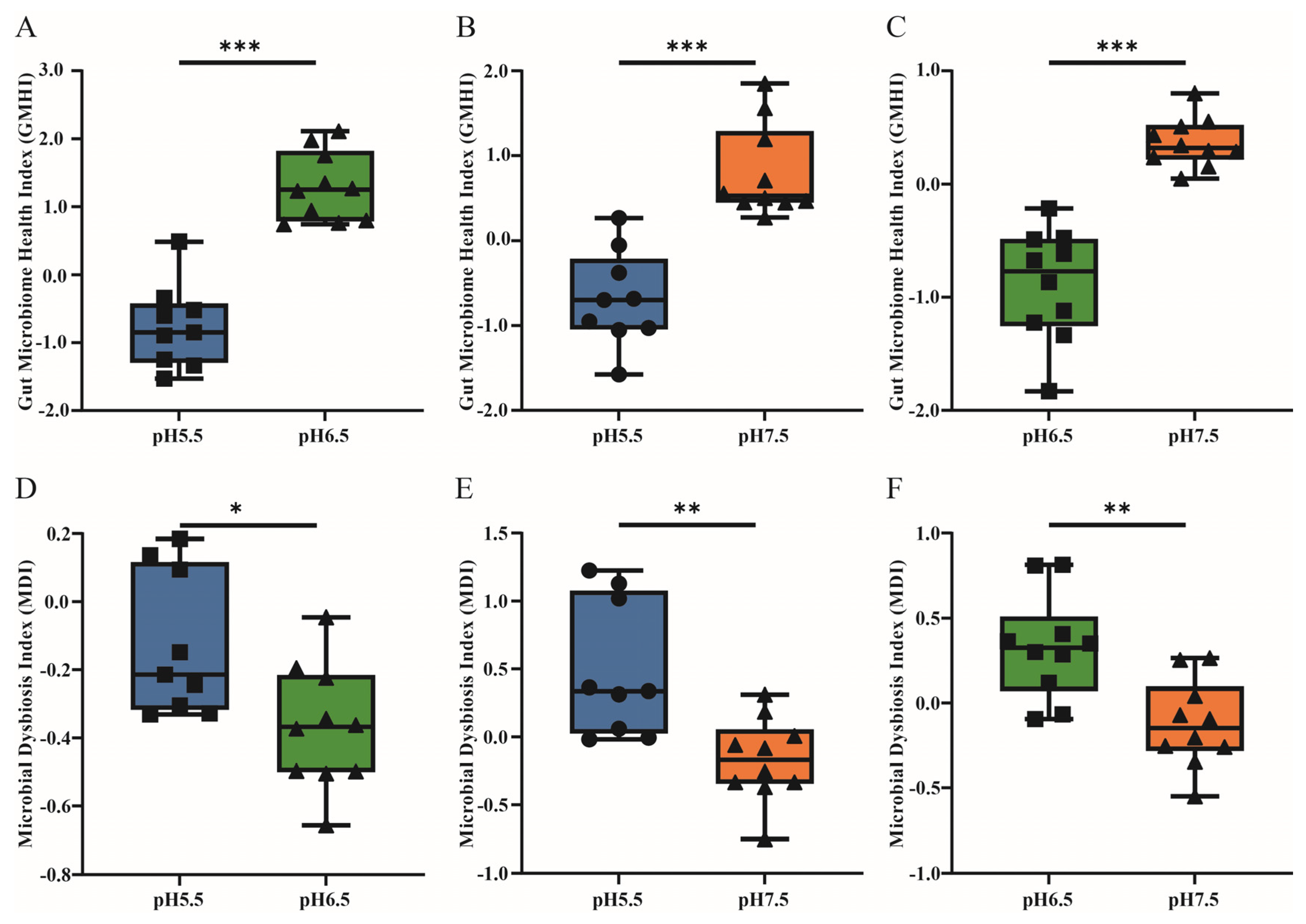
3.3. The Gut Microbiome Health Index and Microbial Dysbiosis Index Analysis
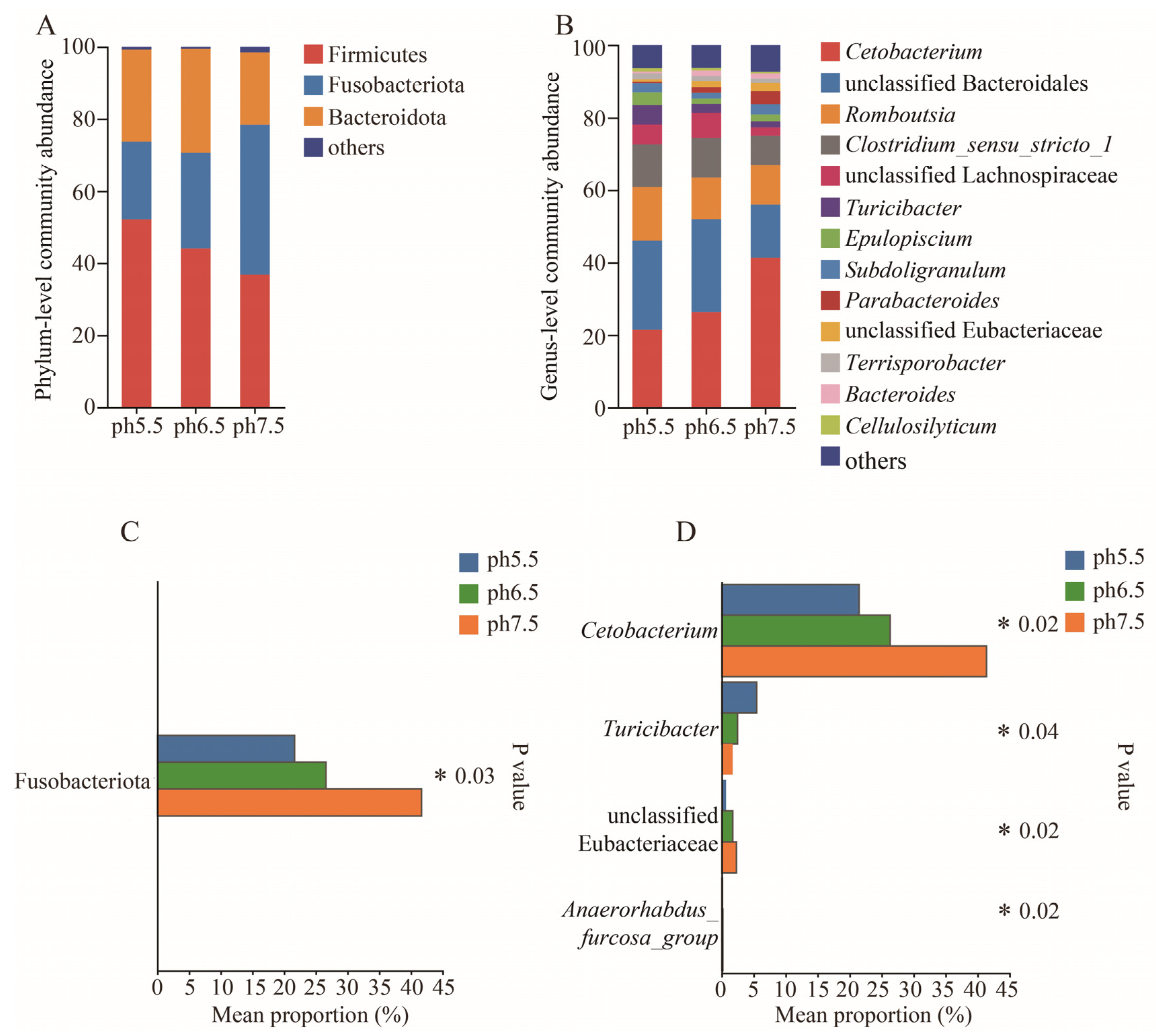
3.4. Colony Composition and Relative Abundance
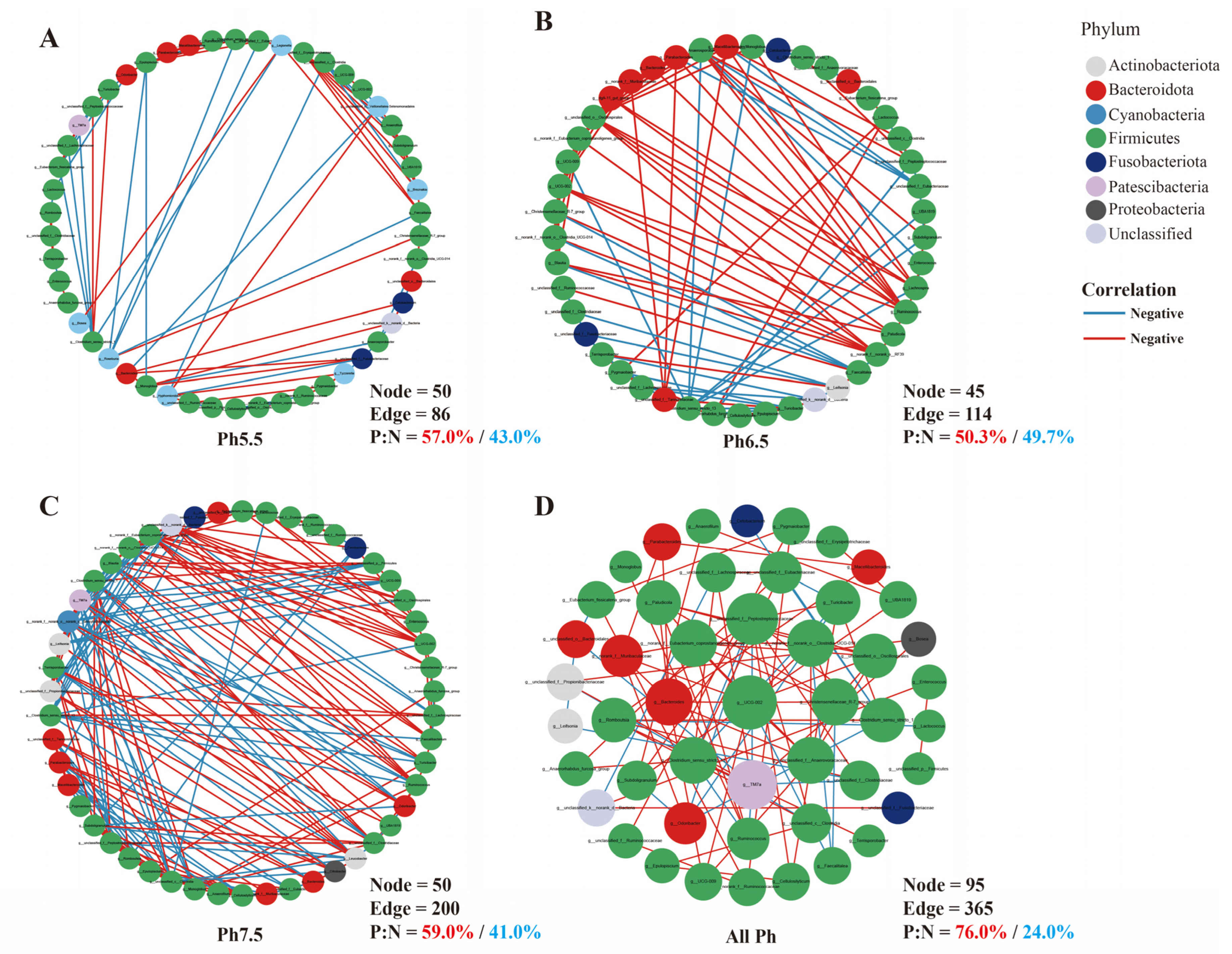
3.5. Gut Microbiota Interaction Network Analysis
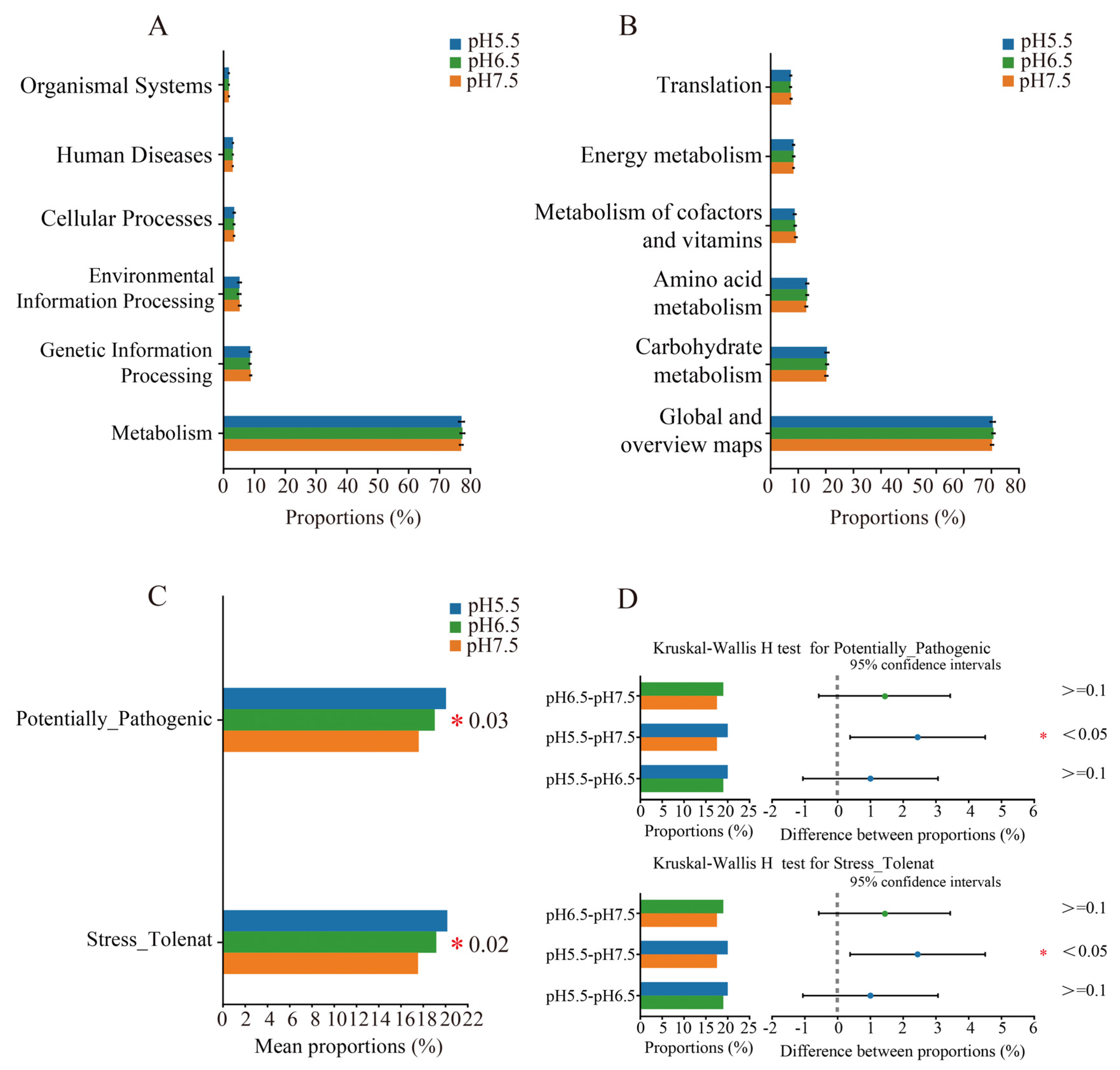
3.6. Predicted Functional Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vézina, A.; Hoegh-Guldberg, O. Effects of ocean acidification on marine ecosystems. Mar. Ecol. Prog. Ser. 2008, 373, 199–201. [Google Scholar] [CrossRef]
- Weiss, L.C.; Pötter, L.; Steiger, A.; Kruppert, S.; Frost, U.; Tollrian, R. Rising pCO2 in freshwater ecosystems has the potential to negatively affect predator-induced defenses in daphnia. Curr. Biol. 2018, 28, 327–332. [Google Scholar] [CrossRef]
- Thomas, A.; Ramkumar, A.; Shanmugam, A. CO2 acidification and its differential responses on aquatic biota—A review. Environ. Adv. 2022, 8, 100219. [Google Scholar] [CrossRef]
- Riebesell, U.; Gattuso, J.P. Lessons Learned from ocean acidification research. Nat. Clim. Change 2015, 5, 12–14. [Google Scholar] [CrossRef]
- Nagelkerken, I.; Goldenberg, S.U.; Ferreira, C.M.; Russell, B.D.; Connell, S.D. Species Interactions drive fish biodiversity loss in a High-CO2 world. Curr. Biol. 2017, 27, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.M.; Frost, T.M.; Ives, A.R. Compensatory dynamics in zooplankton community responses to acidification: Measurement and mechanisms. Ecol. Appl. 2001, 11, 1060–1072. [Google Scholar] [CrossRef]
- Rummer, J.L.; McKenzie, D.J.; Innocenti, A.; Supuran, C.T.; Brauner, C.J. Root effect hemoglobin may have evolved to enhance general tissue oxygen delivery. Science 2013, 340, 1327–1329. [Google Scholar] [CrossRef]
- Ibrahim, A.; Narayanan, M.; Khan, S.; Sheik, M. The effect of freshwater acidification in the tissues of Cyprinus carpio. Glob. J. Zool. 2020, 5, 9–15. [Google Scholar] [CrossRef]
- Horng, J.L.; Kung, G.X.; Lin, L.Y. Acidified water promotes silver-induced toxicity in zebrafish embryos. Aquat. Toxicol. 2024, 268, 106865. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Li, L. Breakeven Point in Ultimate Thickness Between Moisture-Reduced Shrinkage and Thickness Recovery of Densified Softwood Species: Part 1: At Room Temperature. WFS 2018, 50, 373–380. [Google Scholar] [CrossRef]
- Ding, L.; Li, W.; Li, N.; Liang, L.; Zhang, X.; Jin, H.; Shi, H.; Storey, K.B.; Hong, M. Antioxidant responses to salinity stress in an invasive species, the red-eared slider (Trachemys scripta elegans) and involvement of a TOR-Nrf2 signaling pathway. Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 2019, 219, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Huang, Z.; Lu, Y.; Liang, L.; Li, N.; Xu, Z.; Zhang, J.; Shi, H.; Hong, M. Toxic effects of ammonia on intestinal health and microbiota in red-eared slider (Trachemys scripta elegans). Chemosphere 2021, 280, 130630. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Huang, Z.; Ding, L.; Shi, H.; Hong, M. Endoplasmic reticulum unfolded protein response modulates the adaptation of Trachemys scripta elegans in salinity water. Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 2021, 248, 109102. [Google Scholar] [CrossRef]
- Sarkar, A.; McInroy, C.J.A.; Harty, S.; Raulo, A.; Ibata, N.G.O.; Valles-Colomer, M.; Johnson, K.V.-A.; Brito, I.L.; Henrich, J.; Archie, E.A.; et al. Microbial transmission in the social microbiome and host health and disease. Cell 2024, 187, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.; O’Donnell, D.; Jain, N.; Ulrich, J.D.; Herz, J.; Li, Y.; Lemieux, M.; Cheng, J.; Hu, H.; Serrano, J.R.; et al. ApoE isoform– and microbiota-dependent progression of neurodegeneration in a mouse model of tauopathy. Science 2023, 379, eadd1236. [Google Scholar] [CrossRef] [PubMed]
- Begum, N.; Harzandi, A.; Lee, S.; Uhlen, M.; Moyes, D.L.; Shoaie, S. Host-Mycobiome metabolic interactions in health and disease. Gut Microbes 2022, 14, 2121576. [Google Scholar] [CrossRef] [PubMed]
- Forbes, Z.R.; Scro, A.K.; Patel, S.H.; Dourdeville, K.M.; Prescott, R.L.; Smolowitz, R.M. Fecal and cloacal microbiomes of cold-stunned loggerhead Caretta caretta, Kemp’s ridley Lepidochelys kempii, and green sea turtles Chelonia mydas. Endanger. Species. Res. 2023, 50, 93–105. [Google Scholar] [CrossRef]
- Valentino, T.R.; Vechetti, I.J.; Mobley, C.B.; Dungan, C.M.; Golden, L.; Goh, J.; McCarthy, J.J. Dysbiosis of the gut microbiome impairs mouse skeletal muscle adaptation to exercise. J. Physiol. 2021, 599, 4845–4863. [Google Scholar] [CrossRef] [PubMed]
- Al Naggar, Y.; Singavarapu, B.; Paxton, R.J.; Wubet, T. Bees under interactive stressors: The novel insecticides flupyradifurone and sulfoxaflor along with the fungicide azoxystrobin disrupt the gut microbiota of honey bees and increase opportunistic bacterial pathogens. Sci. Total Environ. 2022, 849, 157941. [Google Scholar] [CrossRef]
- Sepulveda, J.; Moeller, A.H. The effects of temperature on animal gut microbiomes. Front. Microbiol. 2020, 11, 512773. [Google Scholar] [CrossRef]
- Soccini, C.; Ferri, V. Bacteriological screening of Trachemys scripta elegans and Emys orbicularis in the Po plain (Italy). Biologia 2004, 59, 201–207. [Google Scholar]
- Du, S. 16S rRNA-Base Analysis of Bacterial Diversity in the Microbial Flora of the Red-Eared Slider and Four Eye-Spotted Turtle Intestinal Tract. Master’s Thesis, Hainan Normal University, Hainan, China, 2015. [Google Scholar]
- Ding, L.; Li, W.; Liang, L.; Huang, Z.; Li, N.; Zhang, J.; Shi, H.; Storey, K.B.; Hong, M. Modulation of the intestinal barrier adaptive functions in red-eared slider (Trachemys scripta elegans) invading brackish waters. Sci. Total. Environ. 2021, 751, 141744. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Huang, Z.; Liang, L.; Shi, H.; Fang, Z.; Hong, M. Comparative toxic effect of ammonia exposure on Mauremys sinensis and invasive species Trachemys scripta elegans. Comp. Biochem. Physiol. C. Toxicol. Pharmacol. 2024, 278, 109847. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhao, D.; Ma, W.; Guo, Y.; Wang, A.; Wang, Q.; Lee, D.J. Denitrifying sulfide removal process on high-salinity wastewaters in the presence of Halomonas sp. Appl. Microbiol. Biotechnol. 2016, 100, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Goebel, B.M. axonomic note: A place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Evol. Microbiol 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. aive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Gupta, V.K.; Kim, M.; Bakshi, U.; Cunningham, K.Y.; Davis, J.M.; Lazaridis, K.N.; Nelson, H.; Chia, N.; Sung, J. A predictive index for health status using species-level gut microbiome profiling. Nat. Commun. 2020, 11, 4635. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Rhoads, W.J.; Edwards, M.A.; Pruden, A. Impact of water heater temperature setting and water use frequency on the building plumbing microbiome. ISME. J. 2017, 11, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Preston, R.; Godoy-Vitorino, F.; Jedlicka, A.; Rodríguez-Hilario, A.; González, H.; Bondy, J.; Lawson, F.; Folawiyo, O.; Michailidi, C.; Dziedzic, A.; et al. 16S rRNA amplicon sequencing identifies microbiota associated with oral cancer, human papilloma virus infection and surgical treatment. Oncotarget 2016, 7, 51320–51334. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and compositionally robust Inference of microbial ecological networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.K.; Yang, R.; Dunitz, J.M.; Boyer, H.C.; Hunter, R.C. 16S rRNA gene sequencing reveals site-specific signatures of the upper and lower airways of cystic fibrosis patients. J. Cyst. Fibros. 2018, 17, 204–212. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Z.; Su, J.; Li, H.; Xiong, J.; Fu, K.; Wang, Z.; Yuan, X.; Shi, Z.; Miao, X.; et al. Altitude-dependent metabolite biomarkers reveal the mechanism of plateau pika adaptation to high altitudes. Integr. Zool. 2023, 18, 1041–1055. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Zhang, X.; Liu, Y.; Chen, H.; Zheng, L.; Zhai, Y.; Zheng, H. The honeybee gut resistome and its role in antibiotic resistance dissemination. Integr. Zool. 2023, 18, 1014–1026. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, B.; Liu, F.; Wang, Z.; Gao, W.; Gan, W.; Chen, H.; Song, Z. Deterministic processes dominate microbial community assembly in artificially bred Schizothorax wangchiachii iuveniles after being released into wild. Integr Zool 2023, 18, 1072–1088. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Z.; Wang, Z.; Wu, Q.; Zhou, J.; Wang, R.; Han, J.; Su, X. omparison of the gut microbiota and metabolism in different regions of Red Swamp Crayfish (Procambarus clarkii). Front. Microbiol. 2023, 14, 1289634. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Han, W.; Cheng, W.; Liu, D.; Wang, W.; Yan, B.; Gao, H.; Hu, G. Impact of ocean acidification on the gut histopathology and intestinal microflora of Exopalaemon carinicauda. Animals 2023, 13, 3299. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Han, S.; Fu, Q.; Yu, Z.; Wang, L.; Song, L. Impact of ocean acidification on the intestinal microflora of the Pacific oyster Crassostrea gigas. Aquaculture 2022, 546, 737365. [Google Scholar] [CrossRef]
- Firrman, J.; Liu, L.; Mahalak, K.; Tanes, C.; Bittinger, K.; Tu, V.; Bobokalonov, J.; Mattei, L.; Zhang, H.; Van den Abbeele, P. The impact of environmental pH on the gut microbiota community structure and short chain fatty acid production. FEMS Microbiol. Ecol. 2022, 98, fiac038. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, F.; Cerqueira, R.; Fuentes, J. Impact of ocean acidification on the intestinal microbiota of the marine sea bream (Sparus aurata L.). Front. Physiol. 2019, 10, 1446. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Fong, J.J.; Parham, J.F.; Shi, H. Reproductive Ecology of the Hainan Four Eye-Spotted Turtle (Sacalia insulensis) on Hainan Island, China. Chelonian Conserv. Biol. 2021, 20, 103–108. [Google Scholar] [CrossRef]
- Xie, M.; Zhou, W.; Xie, Y.; Li, Y.; Zhang, Z.; Yang, Y.; Olsen, R.E.; Ran, C.; Zhou, Z. Effects of Cetobacterium somerae fermentation product on gut and liver health of common carp (Cyprinus Carpio) fed diet supplemented with ultra-micro ground mixed plant proteins. Aquaculture 2021, 543, 736943. [Google Scholar] [CrossRef]
- Xie, M.; Xie, Y.; Li, Y.; Zhou, W.; Zhang, Z.; Yang, Y.; Olsen, R.E.; Ringø, E.; Ran, C.; Zhou, Z. Stabilized fermentation product of Cetobacterium somerae improves gut and liver health and antiviral immunity of zebrafish. Fish Shellfish Immunol. 2022, 120, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhang, Y.; Zhang, Y.; Luo, F.; Song, K.; Wang, G.; Ling, F. Vitamin B12 produced by Cetobacterium somerae improves host resistance against pathogen infection through strengthening the interactions within gut microbiota. Microbiome 2023, 11, 135. [Google Scholar] [CrossRef]
- Li, H.; Cao, W.; Xie, J.; Che, H.; Liu, L.; Dong, X.; Song, L.; Xie, W. α-D-1,6-glucan from Castanea mollissima Blume alleviates dextran sulfate sodium-induced colitis in vivo. Carbohydr. Polym. 2022, 289, 119410. [Google Scholar] [CrossRef]
- Gomes, A.C.; Hoffmann, C.; Mota, J.F. The human gut microbiota: Metabolism and perspective in obesity. Gut Microbes 2018, 9, 308–325. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Lordan, C.; Ross, R.P.; Cotter, P.D. Gut microbes from the phylogenetically diverse genus Eubacterium and their various contributions to gut health. Gut Microbes 2020, 12, 1802866. [Google Scholar] [CrossRef]
- Omori, M.; Maeda, S.; Igarashi, H.; Ohno, K.; Sakai, K.; Yonezawa, T.; Horigome, A.; Odamaki, T.; Matsuki, N. Fecal microbiome in dogs with inflammatory bowel disease and intestinal lymphoma. J. Vet. Med. Sci. 2017, 79, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, Y.; Zhang, J.; Bao, J. Intestinal microbiota in white spot syndrome virus infected red swamp crayfish (Procambarus clarkii) at different health statuses. Aquaculture 2021, 542, 736826. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, Q.; He, Y.; Yu, D.; Chang, G.; Chen, C.; Bi, L.; Lv, J.; Zhao, M.; Lin, X. Associations of gut microbiota and fatty metabolism with immune thrombocytopenia. Front. Med. 2022, 9, 810612. [Google Scholar] [CrossRef] [PubMed]
- Kuziel, G.A.; Rakoff-Nahoum, S. The gut microbiome. Curr. Biol. 2022, 32, 257–264. [Google Scholar] [CrossRef]
- Ahasan, M.S.; Waltzek, T.B.; Huerlimann, R.; Ariel, E. Fecal bacterial communities of wild-captured and stranded green turtles (Chelonia mydas) on the Great Barrier Reef. FEMS Microbiol Ecol. 2017, 93, fix139. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, X.; Dang, Z.; Hong, M.; Shi, H.; Ding, L. Effects of Freshwater Acidification on the Gut Microbial Community of Trachemys scripta elegans. Animals 2024, 14, 1898. https://doi.org/10.3390/ani14131898
Niu X, Dang Z, Hong M, Shi H, Ding L. Effects of Freshwater Acidification on the Gut Microbial Community of Trachemys scripta elegans. Animals. 2024; 14(13):1898. https://doi.org/10.3390/ani14131898
Chicago/Turabian StyleNiu, Xin, Zhaohui Dang, Meiling Hong, Haitao Shi, and Li Ding. 2024. "Effects of Freshwater Acidification on the Gut Microbial Community of Trachemys scripta elegans" Animals 14, no. 13: 1898. https://doi.org/10.3390/ani14131898