
A Pilot Detection and Associate Study of Gene Presence-Absence Variation in Holstein Cattle
 , , , , , and
, , , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. WGS Sequence, Preprocessing, and Alignment
2.2. Phenotypes, dPTA, and Correlation Analysis
2.3. Gene Presence-Absence Variation Identification
2.4. Statistical Overrepresentation Tests of Variable Genes
2.5. Population Genetic Analyses
2.6. Gene PAV-Based GWAS
3. Results
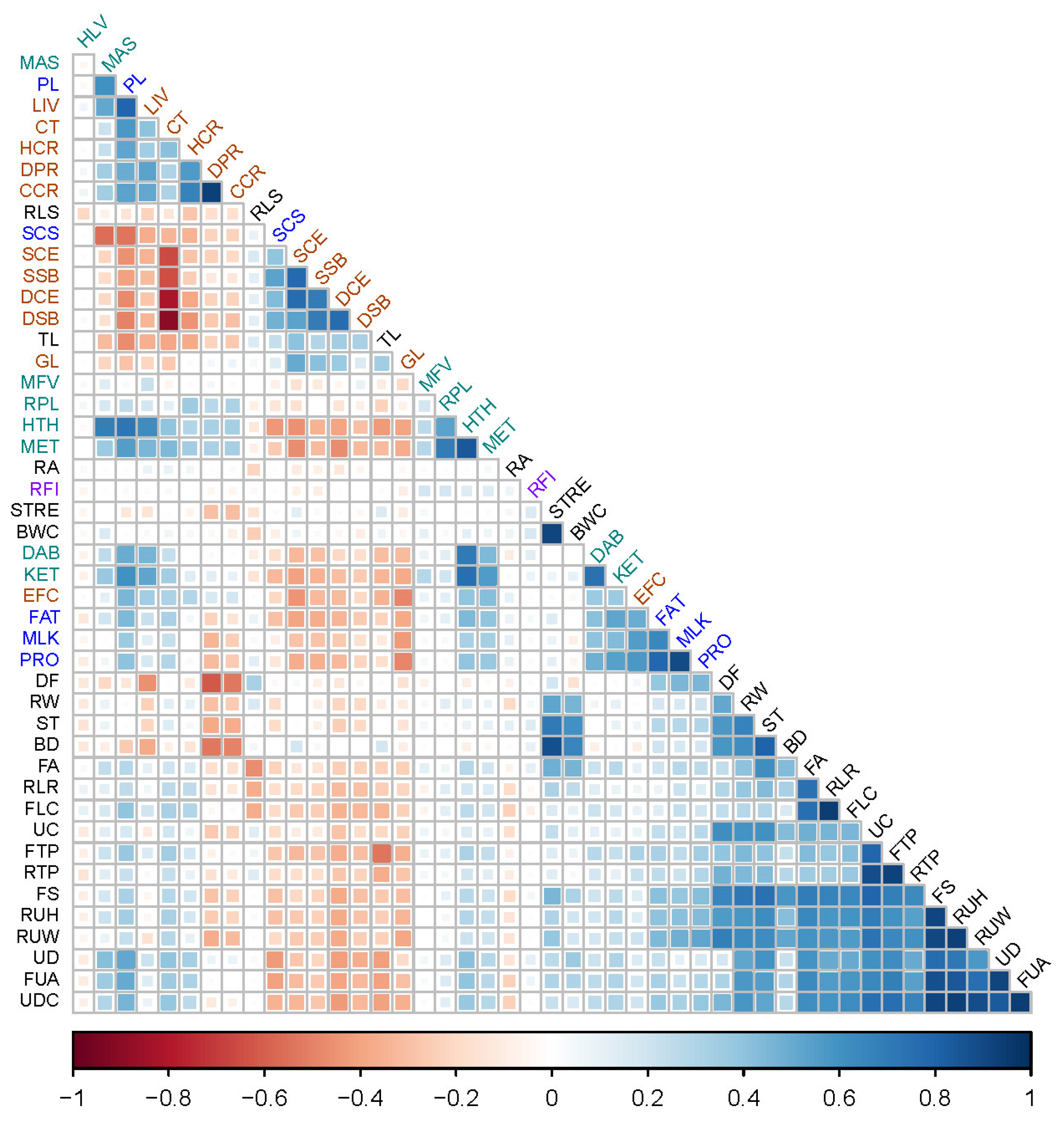
3.1. Phenotype Correlation
3.2. Mapping Reads
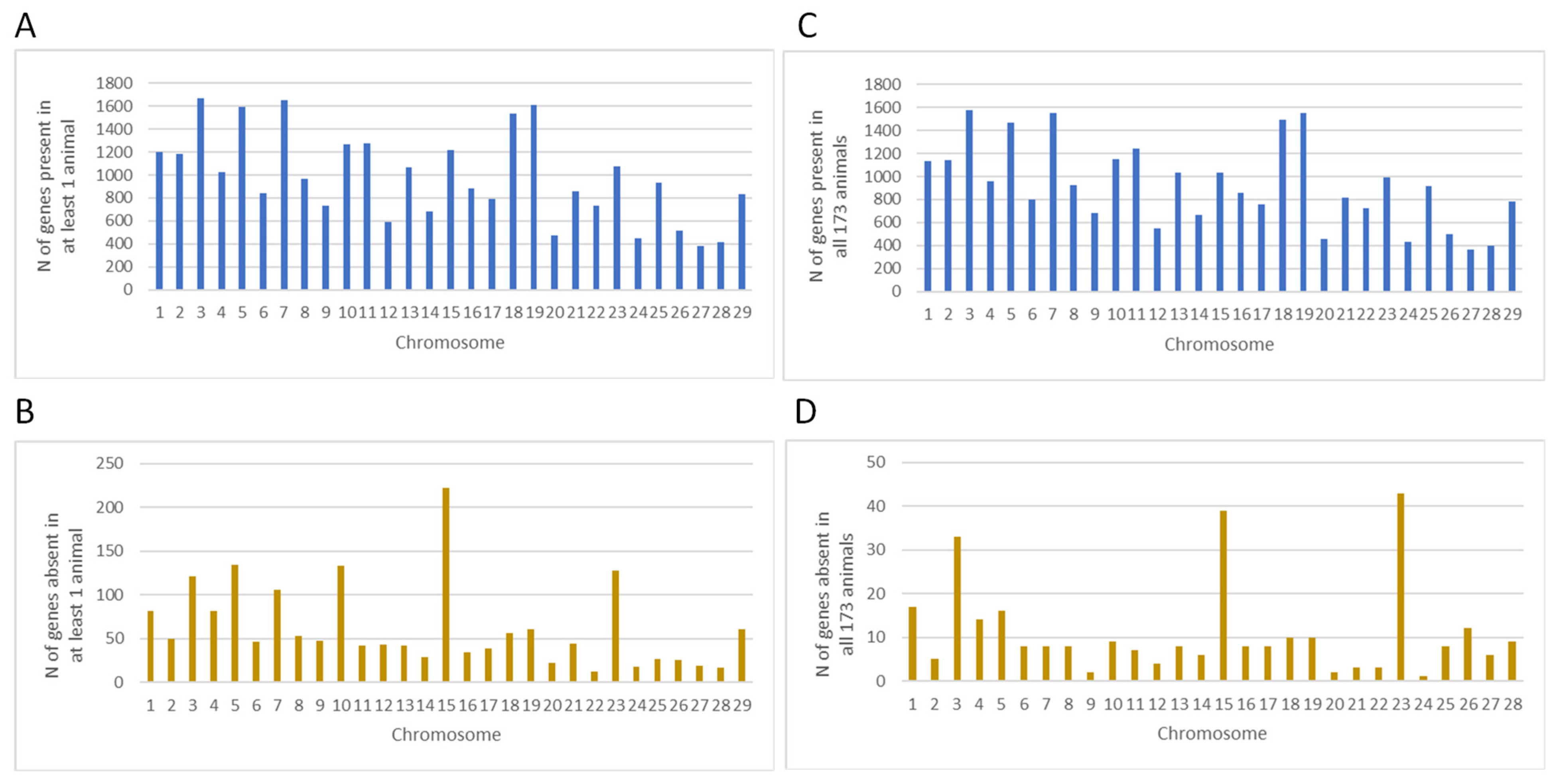
3.3. Identification of Gene PAVs in Holstein Cattle
3.4. PAV Analysis
3.5. Gene PAV-Based GWAS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGPCR | Adhesion G protein-coupled receptors |
| BD | body depth |
| BTA | Bos taurus autosome |
| BWC | body weight composite |
| CCR | cow conception rate |
| CDCB | Council of Dairy Cattle Breeding |
| CNV | copy number variations |
| CT | calving trait composite |
| DAB | displaced abomasum |
| DCE | daughter calving ease |
| DF | dairy form |
| DPR | daughter pregnancy rate |
| dPTA | De-regressed PTA |
| DSB | daughter stillbirth |
| EFC | early first calving |
| FA | foot angle |
| FAT | fat yield |
| FLC | feet and leg composite |
| FS | final score |
| FTP | front teat placement |
| FUA | fore udder attachment |
| GL | gestation length |
| GLM | general linear model |
| GPCR | G protein-coupled receptors |
| GWAS | genome-wide association studies |
| HCR | heifer conception rate |
| HLV | heifer livability |
| HTH | health trait composite |
| INDEL | insertion/deletion |
| JAK | cytokine-activated Janus kinase |
| KET | Ketosis |
| LIV | cow livability |
| MAS | Mastitis |
| MET | Metritis |
| MFV | milk fever/hypocalcemia |
| MLK | milk yield |
| OR | olfactory receptors |
| PAV | Presence-absence variations |
| PCA | principal component analysis |
| PL | productive life |
| PRO | protein yield |
| PTA | transmitting ability |
| RA | rump angle |
| RFI | residual feed intake |
| RLR | rear legs (rear view) |
| RLS | rear legs (side view) |
| RPL | retained placenta |
| RTP | rear teat placement |
| RUH | rear udder height |
| RUW | rear udder width |
| RW | rump width |
| SCE | sire calving ease |
| SCS | somatic cell score |
| SSB | sire stillbirth |
| ST | Stature |
| STAT | signal transducer and activator of transcription |
| STRE | Strength |
| SV | structural variants |
| TL | teat length |
| UC | udder cleft |
| UD | udder depth. |
| UDC | udder composite |
References
- Zimin, A.V.; Delcher, A.L.; Florea, L.; Kelley, D.R.; Schatz, M.C.; Puiu, D.; Hanrahan, F.; Pertea, G.; Van Tassell, C.P.; Sonstegard, T.S.; et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009, 10, R42. [Google Scholar] [CrossRef]
- Weldenegodguad, M.; Popov, R.; Pokharel, K.; Ammosov, I.; Ming, Y.; Ivanova, Z.; Kantanen, J. Whole-Genome Sequencing of Three Native Cattle Breeds Originating From the Northernmost Cattle Farming Regions. Front. Genet. 2019, 9, 728. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, L.; Han, X.; Han, J.; Hu, Y.; Li, F.; Xia, H.; Peng, L.; Boschiero, C.; Rosen, B.D.; et al. Assembly of a pangenome for global cattle reveals missing sequences and novel structural variations, providing new insights into their diversity and evolutionary history. Genome Res. 2022, 32, 1585–1601. [Google Scholar]
- Sun, T.; Pei, S.; Liu, Y.; Hanif, Q.; Xu, H.; Chen, N.; Lei, C.; Yue, X. Whole genome sequencing of simmental cattle for SNP and CNV discovery. BMC Genom. 2023, 24, 179. [Google Scholar]
- Jiang, L.; Kon, T.; Chen, C.; Ichikawa, R.; Zheng, Q.; Pei, L.; Takemura, I.; Nsobi, L.H.; Tabata, H.; Pan, H.; et al. Whole-genome sequencing of endangered Zhoushan cattle suggests its origin and the association of MC1R with black coat colour. Sci. Rep. 2021, 11, 17359. [Google Scholar] [CrossRef]
- Peripolli, E.; Reimer, C.; Ha, N.T.; Geibel, J.; Machado, M.A.; Panetto, J.C.D.C.; do Egito, A.A.; Baldi, F.; Simianer, H.; da Silva, M.V.G.B. Genome-wide detection of signatures of selection in indicine and Brazilian locally adapted taurine cattle breeds using whole-genome re-sequencing data. BMC Genom. 2020, 21, 624. [Google Scholar]
- Paguem, A.; Abanda, B.; Achukwi, M.D.; Baskaran, P.; Czemmel, S.; Renz, A.; Eisenbarth, A. Whole genome characterization of autochthonous Bos taurus brachyceros and introduced Bos indicus indicus cattle breeds in Cameroon regarding their adaptive phenotypic traits and pathogen resistance. BMC Genet. 2020, 21, 64. [Google Scholar]
- Liu, G.E.; Hou, Y.; Zhu, B.; Cardone, M.F.; Jiang, L.; Cellamare, A.; Mitra, A.; Alexander, L.J.; Coutinho, L.L.; Dell’Aquila, M.E.; et al. Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010, 20, 693–703. [Google Scholar]
- da Silva, J.M.; Giachetto, P.F.; da Silva, L.O.; Cintra, L.C.; Paiva, S.R.; Yamagishi, M.E.; Caetano, A.R. Genome-wide copy number variation (CNV) detection in Nelore cattle reveals highly frequent variants in genome regions harboring QTLs affecting production traits. BMC Genom. 2016, 17, 454. [Google Scholar]
- Yang, L.; Xu, L.; Zhu, B.; Niu, H.; Zhang, W.; Miao, J.; Shi, X.; Zhang, M.; Chen, Y.; Zhang, L.; et al. Genome-wide analysis reveals differential selection involved with copy number variation in diverse Chinese Cattle. Sci. Rep. 2017, 7, 14299. [Google Scholar] [CrossRef]
- Lee, Y.L.; Bosse, M.; Mullaart, E.; Groenen, M.A.M.; Veerkamp, R.F.; Bouwman, A.C. Functional and population genetic features of copy number variations in two dairy cattle populations. BMC Genom. 2020, 21, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, L.; Reynolds, E.; Huang, X.; Garrick, D.; Shi, Y. Discovering Copy Number Variation in Dual-Purpose XinJiang Brown Cattle. Front. Genet. 2022, 12, 747431. [Google Scholar] [CrossRef]
- Boussaha, M.; Esquerré, D.; Barbieri, J.; Djari, A.; Pinton, A.; Letaief, R.; Salin, G.; Escudié, F.; Roulet, A.; Fritz, S.; et al. Genome-Wide Study of Structural Variants in Bovine Holstein, Montbéliarde and Normande Dairy Breeds. PLoS ONE 2015, 10, e0135931. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chamberlain, A.J.; Reich, C.M.; Daetwyler, H.D.; Hayes, B.J. Detection and validation of structural variations in bovine whole-genome sequence data. Genet. Sel. Evol. 2017, 49, 13. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ma, L.; Liu, G.E. Initial Analysis of Structural Variation Detections in Cattle Using Long-Read Sequencing Methods. Genes 2022, 13, 828. [Google Scholar] [CrossRef] [PubMed]
- Leonard, A.S.; Crysnanto, D.; Fang, Z.H.; Heaton, M.P.; Vander Ley, B.L.; Herrera, C.; Bollwein, H.; Bickhart, D.M.; Kuhn, K.L.; Smith, T.P.L.; et al. Structural variant-based pangenome construction has low sensitivity to variability of haplotype-resolved bovine assemblies. Nat. Commun. 2022, 13, 3012. [Google Scholar] [CrossRef]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Cheng, Z.; Eichler, E.E. Structural variation of the human genome. Annu. Rev. Genom. Hum. Genet. 2006, 7, 407–442. [Google Scholar] [CrossRef]
- Swanson-Wagner, R.A.; Eichten, S.R.; Kumari, S.; Tiffin, P.; Stein, J.C.; Ware, D.; Springer, N.M. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res. 2010, 20, 1689–1699. [Google Scholar] [CrossRef]
- Marroni, F.; Pinosio, S.; Morgante, M. Structural variation and genome complexity: Is dispensable really dispensable? Curr. Opin. Plant Biol. 2014, 18, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef]
- Sherman, R.M.; Salzberg, S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef]
- Korona, R. Gene dispensability. Curr. Opin. Biotechnol. 2011, 22, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Li, G.; Zhao, H.; Wang, G.; Lian, X.; Xie, W. Exploring the rice dispensable genome using a metagenome-like assembly strategy. Genome Biol. 2015, 16, 187. [Google Scholar] [CrossRef]
- Gerdol, M.; Moreira, R.; Cruz, F.; Gómez-Garrido, J.; Vlasova, A.; Rosani, U.; Venier, P.; Naranjo-Ortiz, M.A.; Murgarella, M.; Greco, S.; et al. Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol. 2020, 21, 275. [Google Scholar] [CrossRef]
- Wang, K.; Hu, H.; Tian, Y.; Li, J.; Scheben, A.; Zhang, C.; Li, Y.; Wu, J.; Yang, L.; Fan, X.; et al. The Chicken Pan-Genome Reveals Gene Content Variation and a Promoter Region Deletion in IGF2BP1 Affecting Body Size. Mol. Biol. Evol. 2021, 38, 5066–5081. [Google Scholar] [CrossRef]
- Li, Z.; Liu, X.; Wang, C.; Li, Z.; Jiang, B.; Zhang, R.; Tong, L.; Qu, Y.; He, S.; Chen, H.; et al. The pig pangenome provides insights into the roles of coding structural variations in genetic diversity and adaptation. Genome Res. 2023, 33, 1833–1847. [Google Scholar] [CrossRef] [PubMed]
- Gabur, I.; Chawla, H.S.; Lopisso, D.T.; von Tiedemann, A.; Snowdon, R.J.; Obermeier, C. Gene presence-absence variation associates with quantitative Verticillium longisporum disease resistance in Brassica napus. Sci. Rep. 2020, 10, 4131. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176.e13. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, C.; Schwaninger, H.; Chao, C.T.; Ma, Y.; Duan, N.; Khan, A.; Ban, S.; Xu, K.; Cheng, L.; et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 2020, 52, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Song, J.M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Saco, A.; Rey-Campos, M.; Gallardo-Escárate, C.; Gerdol, M.; Novoa, B.; Figueras, A. Gene presence/absence variation in Mytilus galloprovincialis and its implications in gene expression and adaptation. iScience 2023, 26, 107827. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Hutchison, J.L.; Null, D.J.; VanRaden, P.M.; Cole, J.B. Reducing animal sequencing redundancy by preferentially selecting animals with low-frequency haplotypes. J. Dairy Sci. 2016, 99, 5526–5534. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 2020, 9, giaa021. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- “Picard Toolkit” Broad Institute, GitHub Repository. 2019. Available online: https://broadinstitute.github.io/picard/ (accessed on 9 February 2023).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef]
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef] [PubMed]
- Tay Fernandez, C.G.; Marsh, J.I.; Nestor, B.J.; Gill, M.; Golicz, A.A.; Bayer, P.E.; Edwards, D. An SGSGeneloss-Based Method for Constructing a Gene Presence-Absence Table Using Mosdepth. Methods Mol. Biol. 2022, 2512, 73–80. [Google Scholar] [PubMed]
- Golicz, A.A.; Martinez, P.A.; Zander, M.; Patel, D.A.; Van De Wouw, A.P.; Visendi, P.; Fitzgerald, T.L.; Edwards, D.; Batley, J. Gene loss in the fungal canola pathogen Leptosphaeria maculans. Funct. Integr. Genom. 2015, 15, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef]
- Blomen, V.A.; Májek, P.; Jae, L.T.; Bigenzahn, J.W.; Nieuwenhuis, J.; Staring, J.; Sacco, R.; van Diemen, F.R.; Olk, N.; Stukalov, A.; et al. Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350, 1092–1096. [Google Scholar] [CrossRef]
- Pons, C.; van Leeuwen, J. Meta-analysis of dispensable essential genes and their interactions with bypass suppressors. Life Sci. Alliance 2023, 7, e202302192. [Google Scholar] [CrossRef] [PubMed]
- Crysnanto, D.; Leonard, A.S.; Fang, Z.H.; Pausch, H. Novel functional sequences uncovered through a bovine multiassembly graph. Proc. Natl. Acad. Sci. USA 2021, 118, e2101056118. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. DAP10- and DAP12-associated receptors in innate immunity. Immunol. Rev. 2009, 227, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.Z.; Khan, A.; Xiao, J.; Ma, Y.; Ma, J.; Gao, J.; Cao, Z. Role of the JAK-STAT Pathway in Bovine Mastitis and Milk Production. Animals 2020, 10, 2107. [Google Scholar] [CrossRef] [PubMed]
- Zozulya, S.; Echeverri, F.; Nguyen, T. The human olfactory receptor repertoire. Genome Biol. 2001, 2, research0018.1. [Google Scholar] [CrossRef] [PubMed]
- Glusman, G.; Yanai, I.; Rubin, I.; Lancet, D. The complete human olfactory subgenome. Genome Res. 2001, 11, 685–702. [Google Scholar] [CrossRef] [PubMed]
- Olender, T.; Lancet, D.; Nebert, D.W. Update on the olfactory receptor (OR) gene superfamily. Hum. Genom. 2008, 3, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Hasin-Brumshtein, Y.; Lancet, D.; Olender, T. Human olfaction: From genomic variation to phenotypic diversity. Trends Genet. 2009, 25, 178–184. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Balasubramanian, S.; Frankish, A.; Huang, N.; Morris, J.; Walter, K.; Jostins, L.; Habegger, L.; Pickrell, J.K.; Montgomery, S.B.; et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012, 335, 823–828, Erratum in Science 2012, 336, 296. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Nguyen, D.T.; Choi, M.; Cha, S.Y.; Kim, J.H.; Dadi, H.; Seo, H.G.; Seo, K.; Chun, T.; Park, C. Analysis of cattle olfactory subgenome: The first detail study on the characteristics of the complete olfactory receptor repertoire of a ruminant. BMC Genom. 2013, 14, 596. [Google Scholar] [CrossRef] [PubMed]
- Trimmer, C.; Keller, A.; Murphy, N.R.; Snyder, L.L.; Willer, J.R.; Nagai, M.H.; Katsanis, N.; Vosshall, L.B.; Matsunami, H.; Mainland, J.D. Genetic variation across the human olfactory receptor repertoire alters odor perception. Proc. Natl. Acad. Sci. USA 2019, 116, 9475–9480. [Google Scholar] [CrossRef]
- Keller, A.; Zhuang, H.; Chi, Q.; Vosshall, L.B.; Matsunami, H. Genetic variation in a human odorant receptor alters odour perception. Nature 2007, 449, 468–472. [Google Scholar] [CrossRef]
- McRae, J.F.; Mainland, J.D.; Jaeger, S.R.; Adipietro, K.A.; Matsunami, H.; Newcomb, R.D. Genetic variation in the odorant receptor OR2J3 is associated with the ability to detect the “grassy” smelling odor, cis-3-hexen-1-ol. Chem. Senses 2012, 37, 585–593. [Google Scholar] [CrossRef]
- Arthur, P.F.; Archer, J.A.; Johnston, D.J.; Herd, R.M.; Richardson, E.C.; Parnell, P.F. Genetic and phenotypic variance and covariance components for feed intake, feed efficiency, and other postweaning traits in Angus cattle. J. Anim. Sci. 2001, 79, 2805–2811. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Connor, E.E.; Wiggans, G.R.; Lu, Y.; Tempelman, R.J.; Schroeder, S.G.; Chen, H.; Liu, G.E. Genome-wide copy number variant analysis reveals variants associated with 10 diverse production traits in Holstein cattle. BMC Genom. 2018, 19, 314. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, S.; Gao, J.; Chen, C.; Zhang, X.; Yuan, H.; Chen, Z.; Yin, X.; Sun, C.; Mao, Y.; et al. Genome-wide analysis of pseudogenes reveals HBBP1’s human-specific essentiality in erythropoiesis and implication in β-thalassemia. Dev. Cell 2021, 56, 478–493.e11. [Google Scholar] [CrossRef]
- Pink, R.C.; Wicks, K.; Caley, D.P.; Punch, E.K.; Jacobs, L.; Carter, D.R. Pseudogenes: Pseudo-functional or key regulators in health and disease? RNA 2011, 17, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Bae, J.; Kim, S.; Lee, S.; Kang, H.; Kim, J.; Bang, I.; Kim, K.; Huh, W.K.; Seok, C.; et al. Understanding the molecular mechanisms of odorant binding and activation of the human OR52 family. Nat. Commun. 2023, 14, 8105. [Google Scholar] [CrossRef] [PubMed]
- Geithe, C.; Andersen, G.; Malki, A.; Krautwurst, D. A Butter Aroma Recombinate Activates Human Class-I Odorant Receptors. J. Agric. Food Chem. 2015, 63, 9410–9420. [Google Scholar] [CrossRef]
- Fleischer, J.; Bumbalo, R.; Bautze, V.; Strotmann, J.; Breer, H. Expression of odorant receptor Olfr78 in enteroendocrine cells of the colon. Cell Tissue Res. 2015, 361, 697–710. [Google Scholar] [CrossRef]
- Julliard, A.K.; Al Koborssy, D.; Fadool, D.A.; Palouzier-Paulignan, B. Nutrient Sensing: Another Chemosensitivity of the Olfactory System. Front. Physiol. 2017, 8, 468. [Google Scholar] [CrossRef]
- Connor, E.E.; Zhou, Y.; Liu, G.E. The essence of appetite: Does olfactory receptor variation play a role? J. Anim. Sci. 2018, 96, 1551–1558. [Google Scholar] [CrossRef]
- Veerkamp, R.F.; Coffey, M.; Berry, D.; de Haas, Y.; Strandberg, E.; Bovenhuis, H.; Calus, M.; Wall, E. Genome-wide associations for feed utilisation complex in primiparous Holstein-Friesian dairy cows from experimental research herds in four European countries. Animal 2012, 6, 1738–1749. [Google Scholar] [CrossRef]
- Ramirez-Diaz, J.; Cenadelli, S.; Bornaghi, V.; Bongioni, G.; Montedoro, S.M.; Achilli, A.; Capelli, C.; Rincon, J.C.; Milanesi, M.; Passamonti, M.M.; et al. Identification of genomic regions associated with total and progressive sperm motility in Italian Holstein bulls. J. Dairy. Sci. 2023, 106, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; Evans, R.D.; Berry, D.P. Reaffirmation of known major genes and the identification of novel candidate genes associated with carcass-related metrics based on whole genome sequence within a large multi-breed cattle population. BMC Genom. 2019, 20, 720. [Google Scholar] [CrossRef] [PubMed]
- Jalil Sarghale, A.; Moradi Shahrebabak, M.; Moradi Shahrebabak, H.; Nejati Javaremi, A.; Saatchi, M.; Khansefid, M.; Miar, Y. Genome-wide association studies for methane emission and ruminal volatile fatty acids using Holstein cattle sequence data. BMC Genet. 2020, 21, 129. [Google Scholar] [CrossRef] [PubMed]
- Gurao, A.; Kashyap, S.K.; Singh, R. β-defensins: An innate defense for bovine mastitis. Vet. World 2017, 10, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Izadpanah, A.; Gallo, R.L. Antimicrobial peptides. J. Am. Acad. Dermatol. 2005, 52 3 Pt 1, 381–390, quiz 391-2. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Schwarzenbacher, H.; Mészáros, G.; Fuerst-Waltl, B.; Fuerst, C.; Sölkner, J.; Penasa, M. On the genomic regions associated with milk lactose in Fleckvieh cattle. J. Dairy Sci. 2019, 102, 10088–10099. [Google Scholar] [CrossRef] [PubMed]
- Atashi, H.; Chen, Y.; Wilmot, H.; Bastin, C.; Vanderick, S.; Hubin, X.; Gengler, N. Single-step genome-wide association analyses for selected infrared-predicted cheese-making traits in Walloon Holstein cows. J. Dairy Sci. 2023, 106, 7816–7831. [Google Scholar] [CrossRef] [PubMed]
- Vassilatis, D.K.; Hohmann, J.G.; Zeng, H.; Li, F.; Ranchalis, J.E.; Mortrud, M.T.; Brown, A.; Rodriguez, S.S.; Weller, J.R.; Wright, A.C.; et al. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903–4908. [Google Scholar] [CrossRef] [PubMed]
- Lala, T.; Hall, R.A. Adhesion G protein-coupled receptors: Structure, signaling, physiology, and pathophysiology. Physiol. Rev. 2022, 102, 1587–1624. [Google Scholar] [CrossRef]
- Matmati, M.; Pouwels, W.; van Bruggen, R.; Jansen, M.; Hoek, R.M.; Verhoeven, A.J.; Hamann, J. The human EGF-TM7 receptor EMR3 is a marker for mature granulocytes. J. Leukoc. Biol. 2007, 81, 440–448. [Google Scholar] [CrossRef]
- Benjamin, A.L.; Green, B.B.; Crooker, B.A.; McKay, S.D.; Kerr, D.E. Differential responsiveness of Holstein and Angus dermal fibroblasts to LPS challenge occurs without major differences in the methylome. BMC Genom. 2016, 17, 258. [Google Scholar] [CrossRef] [PubMed]
- Buaban, S.; Lengnudum, K.; Boonkum, W.; Phakdeedindan, P. Genome-wide association study on milk production and somatic cell score for Thai dairy cattle using weighted single-step approach with random regression test-day model. J. Dairy. Sci. 2022, 105, 468–494. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Position | Gene | Gene Name | Trait and Effect Ranking | p-Value | F-Value |
|---|---|---|---|---|---|---|
| Significant Effect | ||||||
| 7 | 17,361,304 | LOC100337044 | Adhesion G protein-coupled receptor E3 | #1 PL | 5.99 × 10−5 | 17.05 |
| 14 | 1,600,535 | LOC112449566 | Cytochrome P450 11B1, mitochondrial-like | #2 PL | 1.01 × 10−4 | 15.96 |
| 15 | 47,227,535 | OR52E6 | Olfactory receptor family 52 subfamily E member 6 | #1 MET | 2.10 × 10−6 | 24.34 |
| 15 | 47,227,535 | OR52E6 | Olfactory receptor family 52 subfamily E member 6 | #1 FAT | 8.66 × 10−6 | 21.21 |
| 15 | 47,245,425 | OR52N2 | Olfactory receptor family 52 subfamily N member 2 | #1 MLK | 4.41 × 10−6 | 22.69 |
| 15 | 47,245,425 | OR52N2 | Olfactory receptor family 52 subfamily N member 2 | #1 PRO | 1.38 × 10−5 | 20.20 |
| Marginal Effect | ||||||
| 3 | 54,809,083 | LOC785445 | Heterogeneous nuclear ribonucleoprotein A1-like | #1 DCE | 1.90 × 10−4 | 14.63 |
| 10 | 22,785,310 | LOC100296997 | T cell receptor alpha variable 14/delta variable 4-like | #1 LIV | 1.61 × 10−4 | 14.97 |
| 14 | 1,600,535 | LOC112449566 | Cytochrome P450 11B1, mitochondrial-like | #1 FUA | 1.60 × 10−4 | 14.99 |
| 15 | 47,227,535 | OR52E6 | Olfactory receptor family 52 subfamily E member 6 | #1 HTH | 1.05 × 10−4 | 15.86 |
| 15 | 47,227,535 | OR52E6 | Olfactory receptor family 52 subfamily E member 6 | #1 EFC | 1.67 × 10−4 | 14.91 |
| 15 | 47,245,425 | OR52N2 | Olfactory receptor family 52 subfamily N member 2 | #2 MET | 1.12 × 10−4 | 15.73 |
| 15 | 49,043,281 | LOC785207 | Olfactory receptor family 52 subfamily S member 2 | #1 FS | 1.33 × 10−4 | 15.38 |
| 18 | 57,253,674 | LOC112442406 | Zinc finger protein 85-like (withdrawn by NCBI) | #1 KET | 1.61 × 10−4 | 14.98 |
| 19 | 41,468,220 | LOC112442670 | Keratin-associated protein 9-7-like | #1 MFV | 1.12 × 10−4 | 15.74 |
| 20 | 6,875,546 | LOC104975198 | Uncharacterized (withdrawn by NCBI) | #1 CCR | 1.85 × 10−4 | 14.69 |
| 27 | 6,023,993 | LOC789175 | beta-defensin 103B-like | #1 FLC | 1.52 × 10−4 | 15.10 |
| 29 | 27,350,311 | LOC782221 | Olfactory receptor family 8 subfamily B member 1AQ | #1 CT | 2.01 × 10−4 | 14.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boschiero, C.; Neupane, M.; Yang, L.; Schroeder, S.G.; Tuo, W.; Ma, L.; Baldwin, R.L., VI; Van Tassell, C.P.; Liu, G.E. A Pilot Detection and Associate Study of Gene Presence-Absence Variation in Holstein Cattle. Animals 2024, 14, 1921. https://doi.org/10.3390/ani14131921
Boschiero C, Neupane M, Yang L, Schroeder SG, Tuo W, Ma L, Baldwin RL VI, Van Tassell CP, Liu GE. A Pilot Detection and Associate Study of Gene Presence-Absence Variation in Holstein Cattle. Animals. 2024; 14(13):1921. https://doi.org/10.3390/ani14131921
Chicago/Turabian StyleBoschiero, Clarissa, Mahesh Neupane, Liu Yang, Steven G. Schroeder, Wenbin Tuo, Li Ma, Ransom L. Baldwin, VI, Curtis P. Van Tassell, and George E. Liu. 2024. "A Pilot Detection and Associate Study of Gene Presence-Absence Variation in Holstein Cattle" Animals 14, no. 13: 1921. https://doi.org/10.3390/ani14131921
APA StyleBoschiero, C., Neupane, M., Yang, L., Schroeder, S. G., Tuo, W., Ma, L., Baldwin, R. L., VI, Van Tassell, C. P., & Liu, G. E. (2024). A Pilot Detection and Associate Study of Gene Presence-Absence Variation in Holstein Cattle. Animals, 14(13), 1921. https://doi.org/10.3390/ani14131921