
Comparative Study of Bacterial Microbiota Differences in the Rumen and Feces of Xinjiang Brown and Holstein Cattle



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. Milk Production Performances Analysis
2.3. DNA Extraction, PCR, and 16s rRNA Sequencing
2.4. Sequence Data Processing
2.5. Bioinformatic and Statistical Analysis
3. Results and Discussion
3.1. Differences of Milk Production Performances between Xinjiang Brown and Holstein Cattle
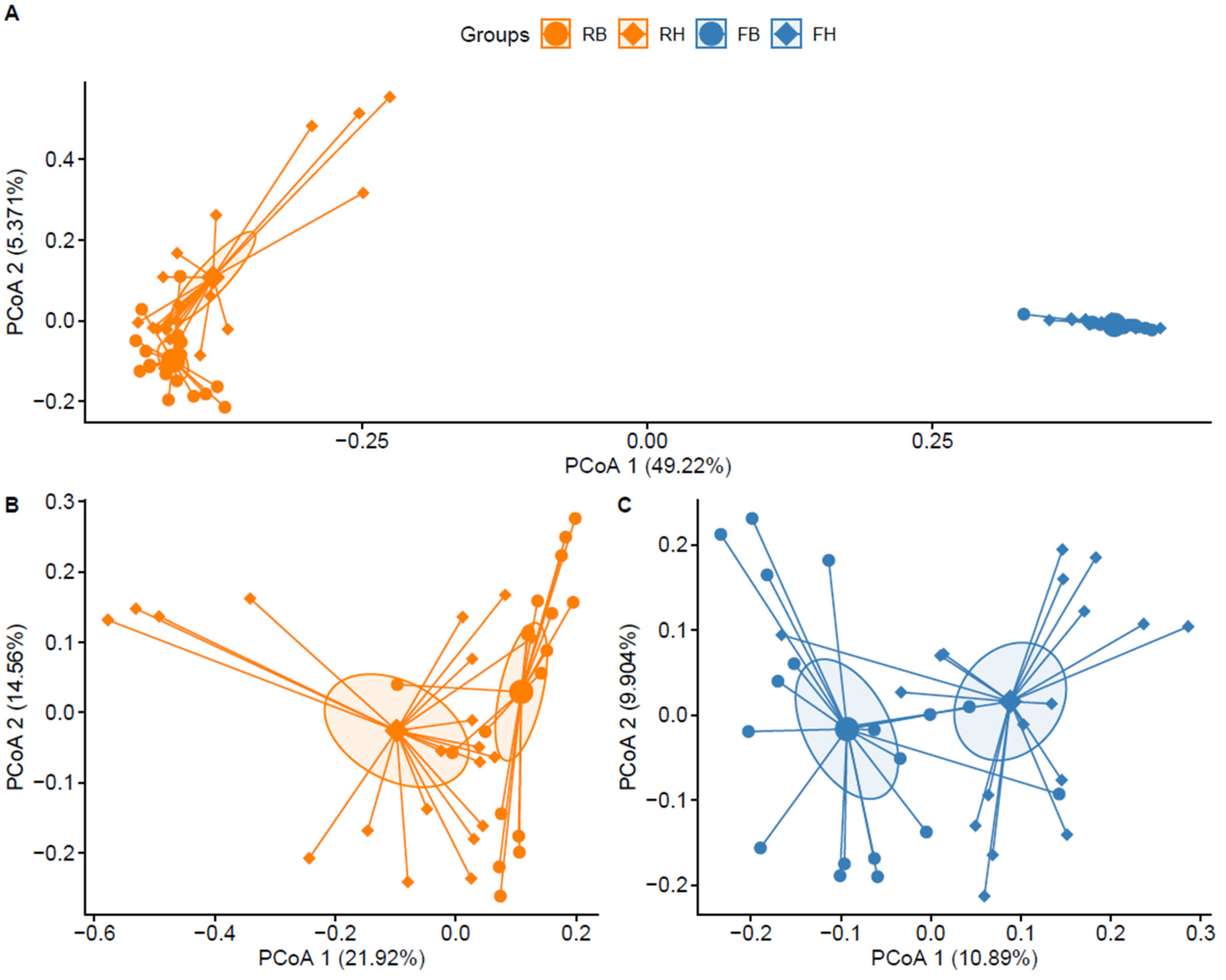
3.2. Microbiota Diversity between Xinjiang Brown and Holstein Cattle
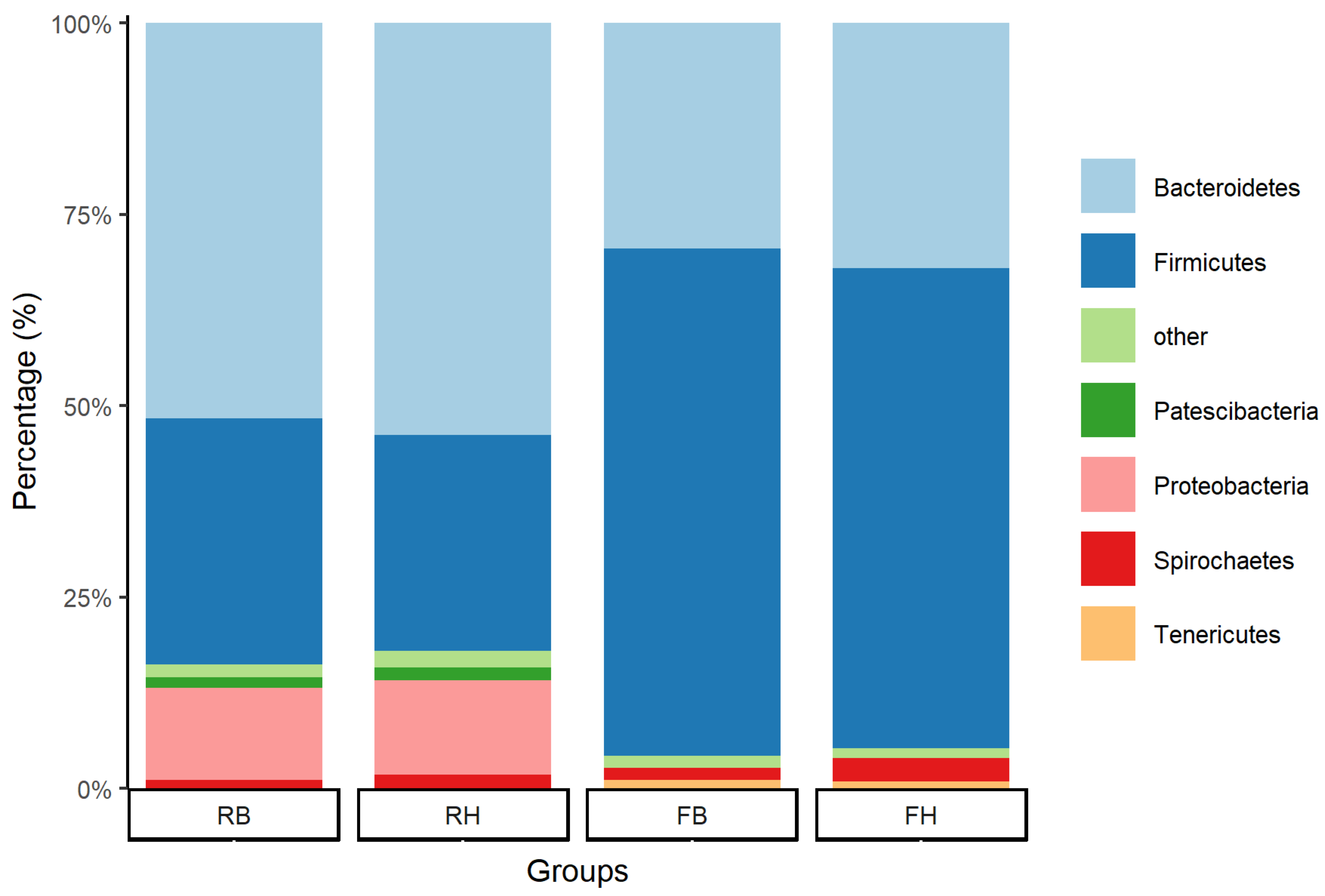
3.3. Relative Abundance of Microbiota at the Phylum Level between Xinjiang Brown and Holstein Cattle
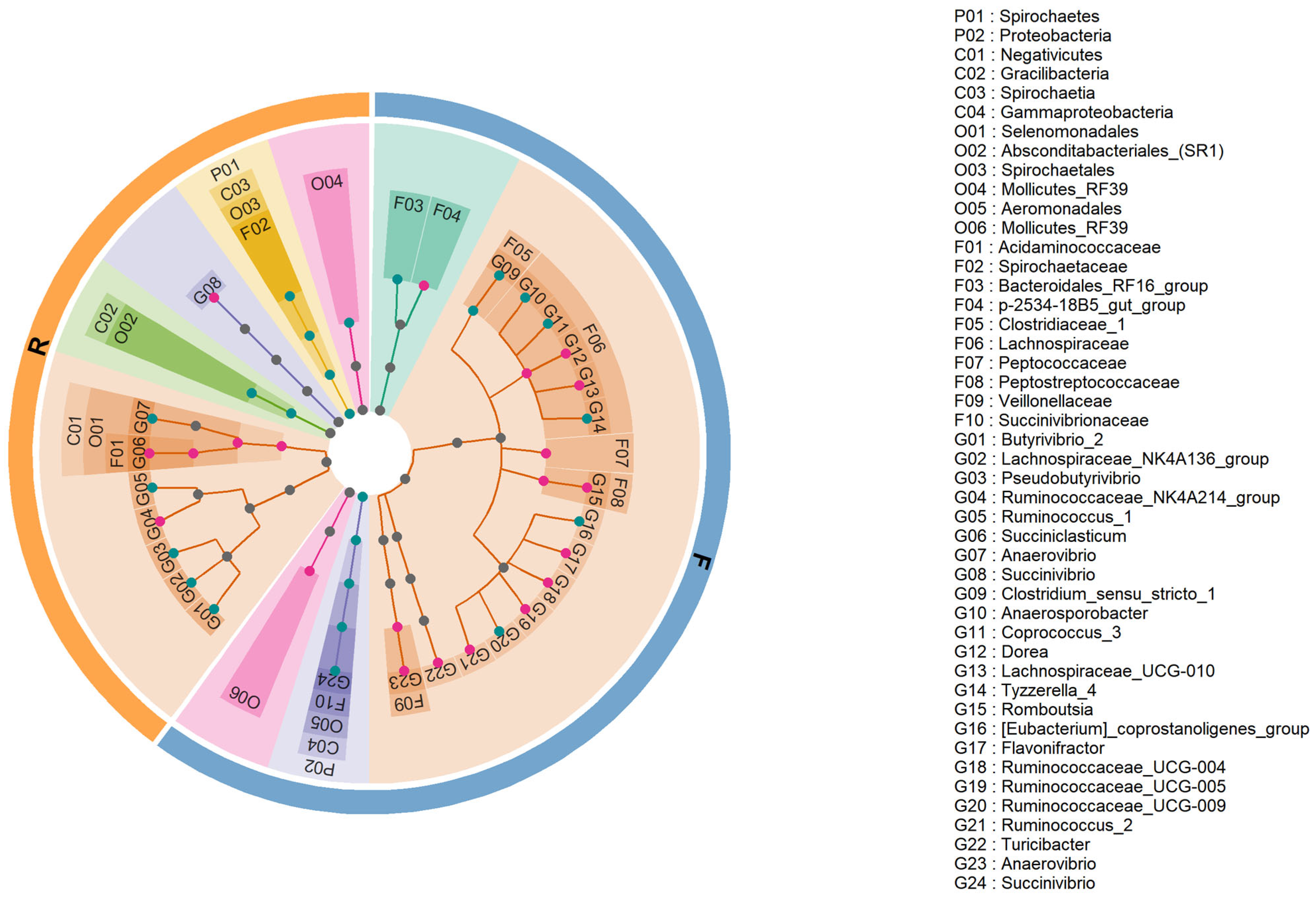
3.4. Significant Differences in Microbiota Composition between Holstein and Xinjiang Brown Cattle
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, L.; Luo, H.; Zhang, X.; Lu, H.; Zhang, M.; Ge, J.; Zhang, T.; Yan, M.; Tan, X.; Huang, X.; et al. Factor Analysis of Genetic Parameters for Body Conformation Traits in Dual-Purpose Simmental Cattle. Animals 2022, 12, 2433. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, L.; Chen, C.J.; Zhang, M.; Lu, X.; Zhang, Z.; Huang, X.; Shi, Y. Genome-Wide Association Study of Milk and Reproductive Traits in Dual-Purpose Xinjiang Brown Cattle. BMC Genom. 2019, 20, 827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Luo, H.; Xu, L.; Shi, Y.; Zhou, J.; Wang, D.; Zhang, X.; Huang, X.; Wang, Y. Genomic Selection for Milk Production Traits in Xinjiang Brown Cattle. Animals 2022, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, Y.; Huang, G.; Zheng, N.; Zhao, S.; Wang, J. Ruminal Bacterial Community Is Associated with the Variations of Total Milk Solid Content in Holstein Lactating Cows. Anim. Nutr. 2022, 9, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Berthiaume, R.; Benchaar, C.; Chaves, A.V.; Tremblay, G.F.; Castonguay, Y.; Bertrand, A.; Bélanger, G.; Michaud, R.; Lafrenière, C.; McAllister, T.A.; et al. Effects of Nonstructural Carbohydrate Concentration in Alfalfa on Fermentation and Microbial Protein Synthesis in Continuous Culture1. J. Dairy Sci. 2010, 93, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.-Y.; Xie, Y.-Y.; Zhong, Y.; Ma, X.-J.; Sun, H.-Z.; Liu, J.-X. Integrated Meta-Omics Reveals New Ruminal Microbial Features Associated with Feed Efficiency in Dairy Cattle. Microbiome 2022, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; White, B.A.; Mizrahi, I. Potential Role of the Bovine Rumen Microbiome in Modulating Milk Composition and Feed Efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef]
- Bainbridge, M.L.; Cersosimo, L.M.; Wright, A.-D.G.; Kraft, J. Rumen Bacterial Communities Shift across a Lactation in Holstein, Jersey and Holstein × Jersey Dairy Cows and Correlate to Rumen Function, Bacterial Fatty Acid Composition and Production Parameters. FEMS Microbiol. Ecol. 2016, 92, fiw059. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.S.; Oikonomou, G.; Lima, S.F.; Bicalho, M.L.S.; Ganda, E.K.; de Oliveira Filho, J.C.; Lorenzo, G.; Trojacanec, P.; Bicalho, R.C. Prepartum and Postpartum Rumen Fluid Microbiomes: Characterization and Correlation with Production Traits in Dairy Cows. Appl. Environ. Microbiol. 2015, 81, 1327–1337. [Google Scholar] [CrossRef]
- Shabat, S.K.B.; Sasson, G.; Doron-Faigenboim, A.; Durman, T.; Yaacoby, S.; Berg Miller, M.E.; White, B.A.; Shterzer, N.; Mizrahi, I. Specific Microbiome-Dependent Mechanisms Underlie the Energy Harvest Efficiency of Ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef]
- Zhao, S.; Min, L.; Zheng, N.; Wang, J. Effect of Heat Stress on Bacterial Composition and Metabolism in the Rumen of Lactating Dairy Cows. Animals 2019, 9, 925. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, C.; Huo, D.; Hu, Q.; Peng, Q. Comparative Study of the Gut Microbiome Potentially Related to Milk Protein in Murrah Buffaloes (Bubalus Bubalis) and Chinese Holstein Cattle. Sci. Rep. 2017, 7, 42189. [Google Scholar] [CrossRef]
- Paz, H.A.; Anderson, C.L.; Muller, M.J.; Kononoff, P.J.; Fernando, S.C. Rumen Bacterial Community Composition in Holstein and Jersey Cows Is Different under Same Dietary Condition and Is Not Affected by Sampling Method. Front. Microbiol. 2016, 7, 206435. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing Taxonomic Classification of Marker-Gene Amplicon Sequences with QIIME 2’s Q2-Feature-Classifier Plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Littell, R.C.; Milliken, G.A.; Stroup, W.W.; Wolfinger, R.D. SASTM System for Mixed Models. Technometrics 1997, 39, 344. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Anderson, M.J. A New Method for Non-Parametric Multivariate Analysis of Variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Xu, S.; Yu, G. MicrobiotaProcess: A Comprehensive R Package for Managing and Analyzing Microbiome and Other Ecological Data within the Tidy Framework. Methods Brief Commun. 2023. [Google Scholar] [CrossRef]
- Wang, Y.; Devkota, S.; Musch, M.W.; Jabri, B.; Nagler, C.; Antonopoulos, D.A.; Chervonsky, A.; Chang, E.B. Regional Mucosa-Associated Microbiota Determine Physiological Expression of TLR2 and TLR4 in Murine Colon. PLoS ONE 2010, 5, e13607. [Google Scholar] [CrossRef] [PubMed]
- Costello, E.K.; Lauber, C.L.; Hamady, M.; Fierer, N.; Gordon, J.I.; Knight, R. Bacterial Community Variation in Human Body Habitats Across Space and Time. Science 2009, 326, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, Y.; Luo, H.; Qiu, W.; Zhang, H.; Hu, L.; Wang, Y.; Dong, G.; Guo, G. The Association Between Inflammaging and Age-Related Changes in the Ruminal and Fecal Microbiota Among Lactating Holstein Cows. Front. Microbiol. 2019, 10, 1803. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, H.; Hu, L.; Zhang, G.; Lu, H.; Luo, H.; Zhao, S.; Zhu, H.; Wang, Y. Characterization of the Microbial Communities along the Gastrointestinal Tract in Crossbred Cattle. Animals 2022, 12, 825. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Vitorino, F.; Goldfarb, K.C.; Karaoz, U.; Leal, S.; Garcia-Amado, M.A.; Hugenholtz, P.; Tringe, S.G.; Brodie, E.L.; Dominguez-Bello, M.G. Comparative Analyses of Foregut and Hindgut Bacterial Communities in Hoatzins and Cows. ISME J. 2012, 6, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Rehman, H.U.; Vahjen, W.; Awad, W.A.; Zentek, J. Indigenous Bacteria and Bacterial Metabolic Products in the Gastrointestinal Tract of Broiler Chickens. Arch. Anim. Nutr. 2007, 61, 319–335. [Google Scholar] [CrossRef]
- Deloris Alexander, A.; Orcutt, R.P.; Henry, J.C.; Baker, J.; Bissahoyo, A.C.; Threadgill, D.W. Quantitative PCR Assays for Mouse Enteric Flora Reveal Strain-Dependent Differences in Composition That Are Influenced by the Microenvironment. Mamm. Genome 2006, 17, 1093–1104. [Google Scholar] [CrossRef]
- Kovacs, A.; Ben-Jacob, N.; Tayem, H.; Halperin, E.; Iraqi, F.A.; Gophna, U. Genotype Is a Stronger Determinant than Sex of the Mouse Gut Microbiota. Microb. Ecol. 2011, 61, 423–428. [Google Scholar] [CrossRef]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in Gut Microbiota Composition Is a Complex Polygenic Trait Shaped by Multiple Environmental and Host Genetic Factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef]
- McKnite, A.M.; Perez-Munoz, M.E.; Lu, L.; Williams, E.G.; Brewer, S.; Andreux, P.A.; Bastiaansen, J.W.M.; Wang, X.; Kachman, S.D.; Auwerx, J.; et al. Murine Gut Microbiota Is Defined by Host Genetics and Modulates Variation of Metabolic Traits. PLoS ONE 2012, 7, e39191. [Google Scholar] [CrossRef]
- Ozbayram, E.; Ince, O.; Ince, B.; Harms, H.; Kleinsteuber, S. Comparison of Rumen and Manure Microbiomes and Implications for the Inoculation of Anaerobic Digesters. Microorganisms 2018, 6, 15. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, M.; Zhang, R.; Zhu, W.; Mao, S. Comparative Studies of the Composition of Bacterial Microbiota Associated with the Ruminal Content, Ruminal Epithelium and in the Feces of Lactating Dairy Cows. Microb. Biotechnol. 2016, 9, 257–268. [Google Scholar] [CrossRef]
- Xue, M.; Sun, H.; Wu, X.; Guan, L.L.; Liu, J. Assessment of Rumen Microbiota from a Large Dairy Cattle Cohort Reveals the Pan and Core Bacteriomes Contributing to Varied Phenotypes. Appl. Environ. Microbiol 2018, 84, e00970-18. [Google Scholar] [CrossRef]
- Boaro, A.A.; Kim, Y.-M.; Konopka, A.E.; Callister, S.J.; Ahring, B.K. Integrated ‘omics Analysis for Studying the Microbial Community Response to a pH Perturbation of a Cellulose-Degrading Bioreactor Culture. FEMS Microbiol. Ecol. 2014, 90, 802–815. [Google Scholar] [CrossRef]
- Liu, C.; Li, X.H.; Chen, Y.X.; Cheng, Z.H.; Duan, Q.H.; Meng, Q.H.; Tao, X.P.; Shang, B.; Dong, H.M. Age-Related Response of Rumen Microbiota to Mineral Salt and Effects of Their Interactions on Enteric Methane Emissions in Cattle. Microb. Ecol. 2017, 73, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Xue, F.; Nan, X.; Tang, Z.; Wang, K.; Beckers, Y.; Jiang, L.; Xiong, B. Illumina Sequencing Approach to Characterize Thiamine Metabolism Related Bacteria and the Impacts of Thiamine Supplementation on Ruminal Microbiota in Dairy Cows Fed High-Grain Diets. Front. Microbiol. 2017, 8, 1818. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Zhang, H.; Yang, D.; Zhang, Y.; Xiong, B.; Jiang, L. Illumina Sequencing Analysis of the Ruminal Microbiota in High-Yield and Low-Yield Lactating Dairy Cows. PLoS ONE 2018, 13, e0198225. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.Y.; Sun, H.Z.; Wu, X.H.; Guan, L.L.; Liu, J.X. Assessment of Rumen Bacteria in Dairy Cows with Varied Milk Protein Yield. J. Dairy Sci. 2019, 102, 5031–5041. [Google Scholar] [CrossRef]
- Mu, Y.; Lin, X.; Wang, Z.; Hou, Q.; Wang, Y.; Hu, Z. High-production Dairy Cattle Exhibit Different Rumen and Fecal Bacterial Community and Rumen Metabolite Profile than Low-production Cattle. MicrobiologyOpen 2019, 8, e00673. [Google Scholar] [CrossRef]
- Guo, B.; Li, D.; Zhou, B.; Jiang, Y.; Bai, H.; Zhang, Y.; Xu, Q.; Zhao, W.; Chen, G. Comparative Characterization of Bacterial Communities in Geese Consuming of Different Proportions of Ryegrass. PLoS ONE 2019, 14, e0223445. [Google Scholar] [CrossRef] [PubMed]
- Ze, X.; Ben David, Y.; Laverde-Gomez, J.A.; Dassa, B.; Sheridan, P.O.; Duncan, S.H.; Louis, P.; Henrissat, B.; Juge, N.; Koropatkin, N.M.; et al. Unique Organization of Extracellular Amylases into Amylosomes in the Resistant Starch-Utilizing Human Colonic Firmicutes Bacterium Ruminococcus Bromii. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Thoetkiattikul, H.; Mhuantong, W.; Laothanachareon, T.; Tangphatsornruang, S.; Pattarajinda, V.; Eurwilaichitr, L.; Champreda, V. Comparative Analysis of Microbial Profiles in Cow Rumen Fed with Different Dietary Fiber by Tagged 16S rRNA Gene Pyrosequencing. Curr. Microbiol. 2013, 67, 130–137. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Item | Least Squares Mean ± SE | p-Value | |
|---|---|---|---|
| Xinjiang Brown | Holstein | ||
| DMY | 18.71 ± 1.22 | 24.68 ± 1.25 | 0.002 |
| Fat | 4.94 ± 0.16 | 3.66 ± 0.16 | <0.001 |
| Protein | 3.91 ± 0.07 | 3.49 ± 0.07 | <0.001 |
| Solids | 14.40 ± 0.16 | 12.79 ± 0.17 | <0.001 |
| Car | 19.29 ± 0.70 | 11.81 ± 0.71 | <0.001 |
| FPD | 540.55 ± 1.90 | 524.46 ± 1.95 | <0.001 |
| SCS | 820.00 ± 206.35 | 451.42 ± 211.82 | 0.217 |
| Lactose | 5.03 ± 0.03 | 5.03 ± 0.03 | 0.977 |
| BHB | 0.87 ± 0.09 | 0.87 ± 0.09 | 0.979 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, H.; Chen, S.; Li, F.; Zhang, G.; Geng, J.; Zhang, M.; Huang, X.; Wang, Y. Comparative Study of Bacterial Microbiota Differences in the Rumen and Feces of Xinjiang Brown and Holstein Cattle. Animals 2024, 14, 1748. https://doi.org/10.3390/ani14121748
Lu H, Chen S, Li F, Zhang G, Geng J, Zhang M, Huang X, Wang Y. Comparative Study of Bacterial Microbiota Differences in the Rumen and Feces of Xinjiang Brown and Holstein Cattle. Animals. 2024; 14(12):1748. https://doi.org/10.3390/ani14121748
Chicago/Turabian StyleLu, Haibo, Shaokan Chen, Fengjie Li, Guoxing Zhang, Juan Geng, Menghua Zhang, Xixia Huang, and Yachun Wang. 2024. "Comparative Study of Bacterial Microbiota Differences in the Rumen and Feces of Xinjiang Brown and Holstein Cattle" Animals 14, no. 12: 1748. https://doi.org/10.3390/ani14121748
APA StyleLu, H., Chen, S., Li, F., Zhang, G., Geng, J., Zhang, M., Huang, X., & Wang, Y. (2024). Comparative Study of Bacterial Microbiota Differences in the Rumen and Feces of Xinjiang Brown and Holstein Cattle. Animals, 14(12), 1748. https://doi.org/10.3390/ani14121748