
Ere, a Family of Short Interspersed Elements in the Genomes of Odd-Toed Ungulates (Perissodactyla)
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatics Methods
2.2. Plasmid Constructs
2.3. Cell Transfection and Northern Blot Analysis
3. Results
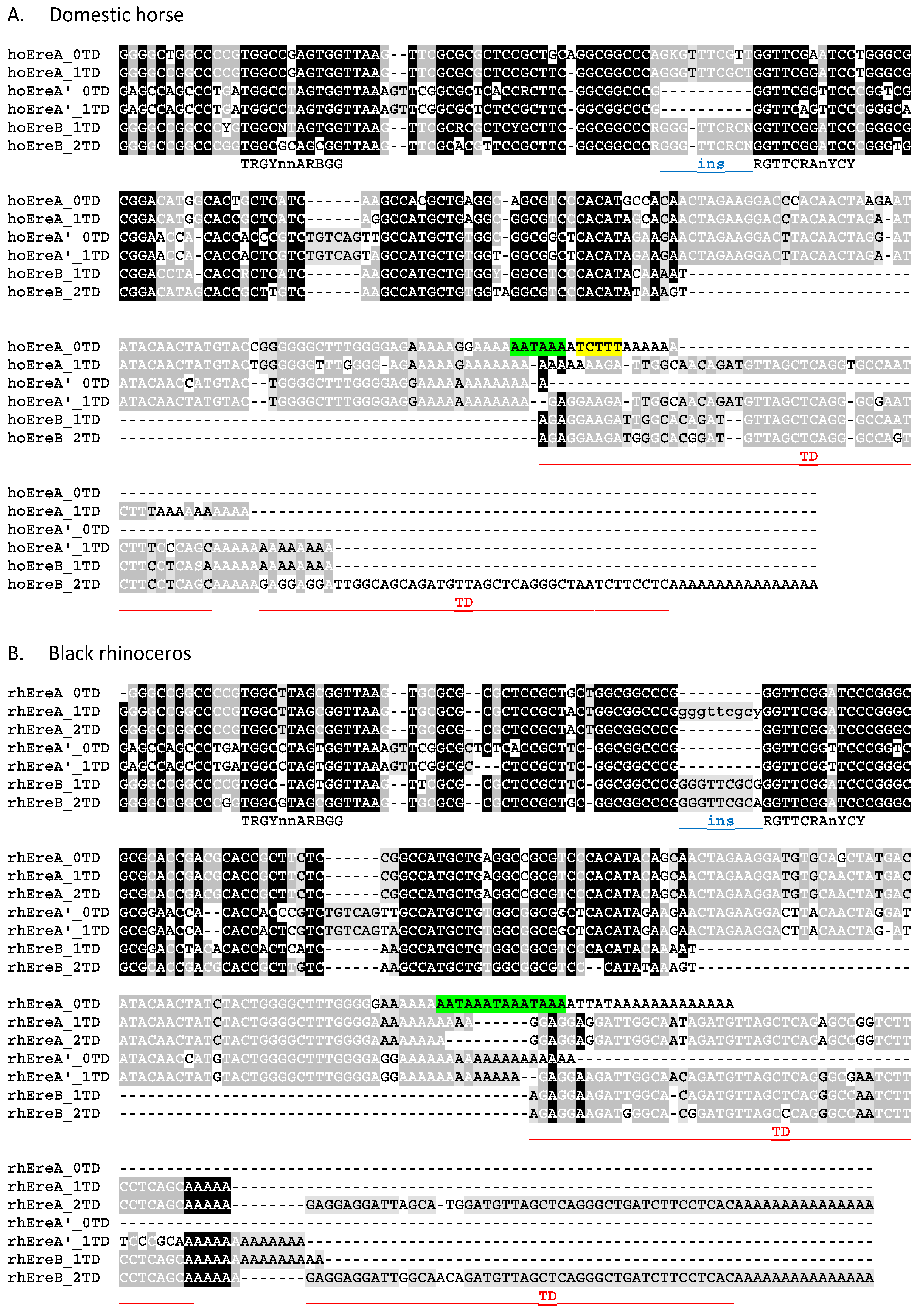
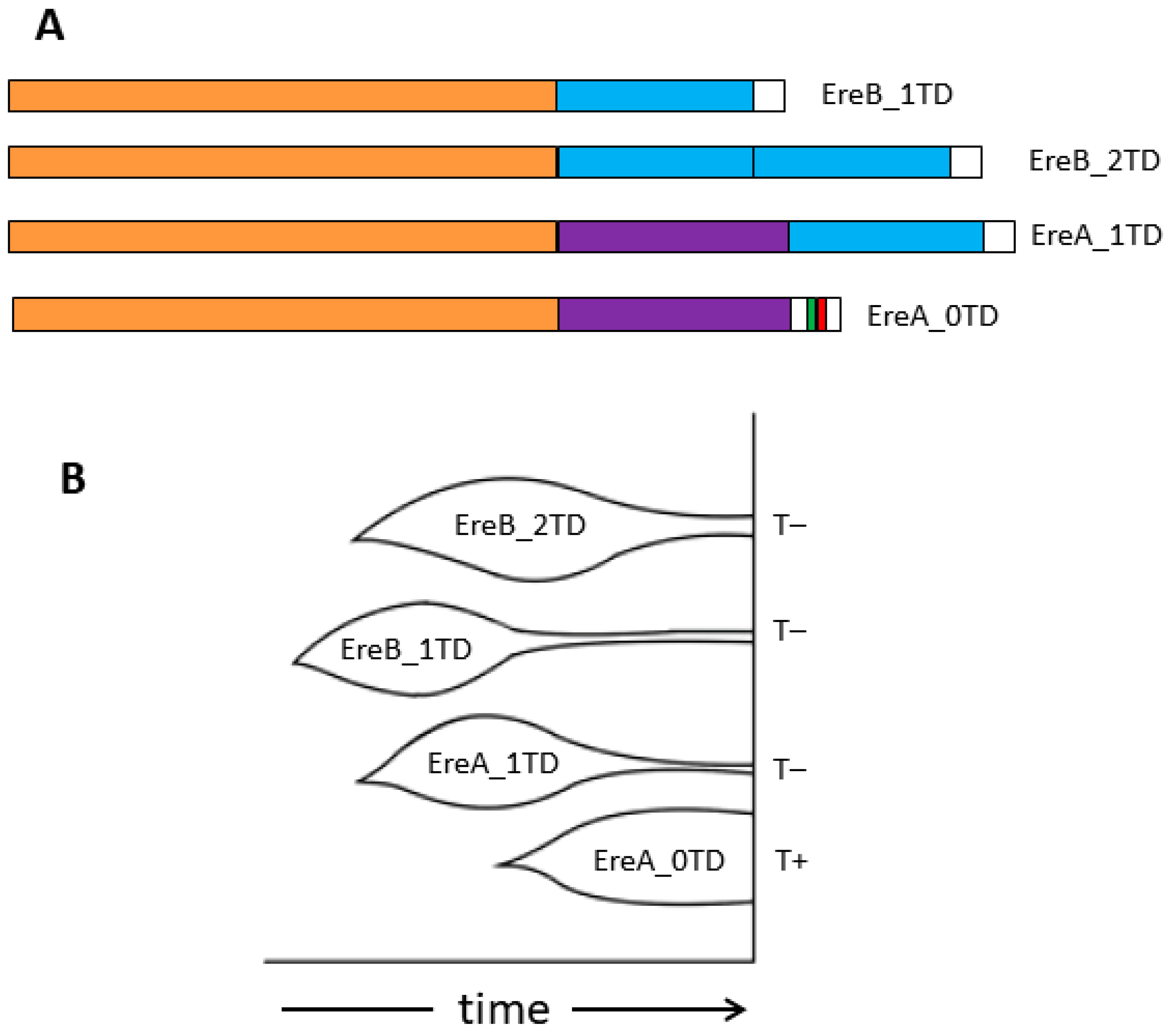
3.1. Structure and Subfamilies of Ere
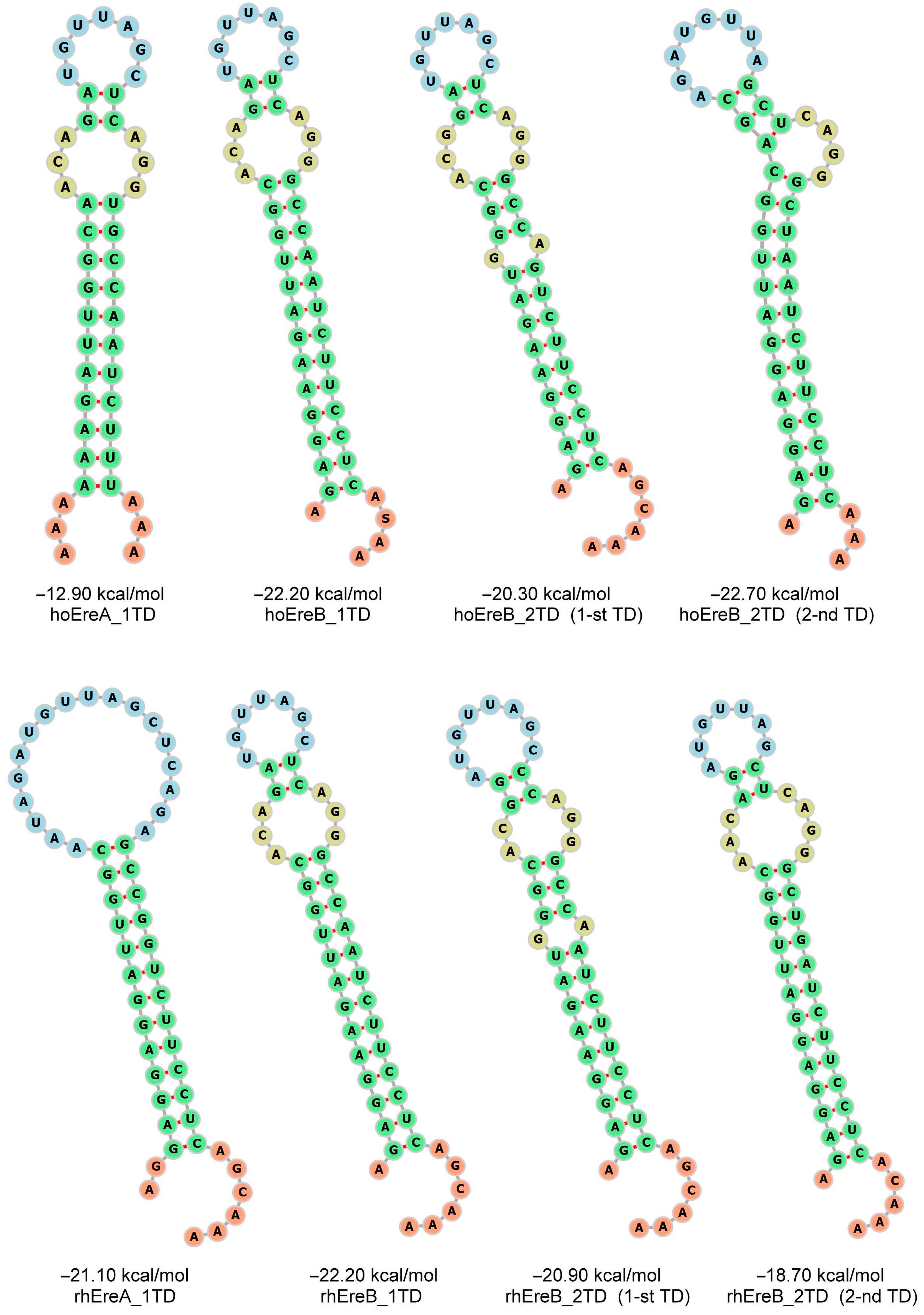
3.2. Ere Subfamilies of T+ SINEs
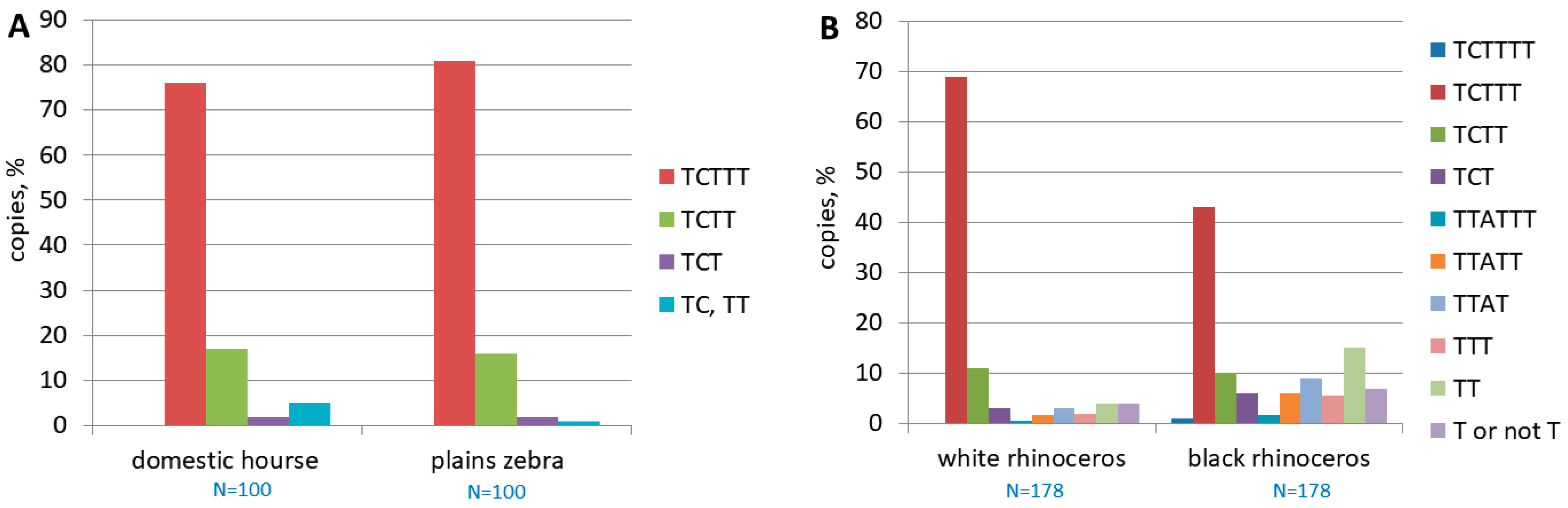
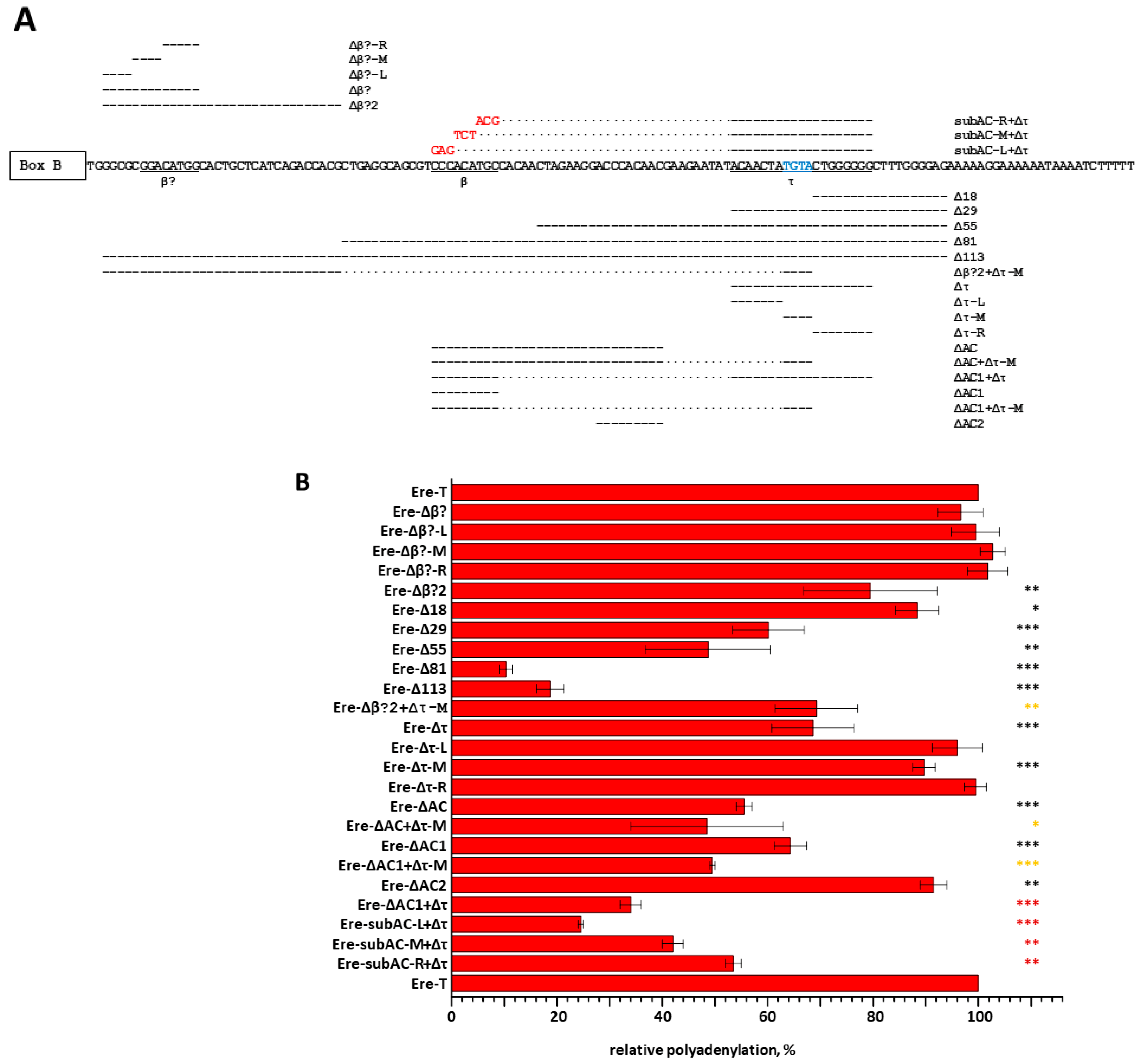
3.3. An Assessment of the Activity of Ere Subfamilies
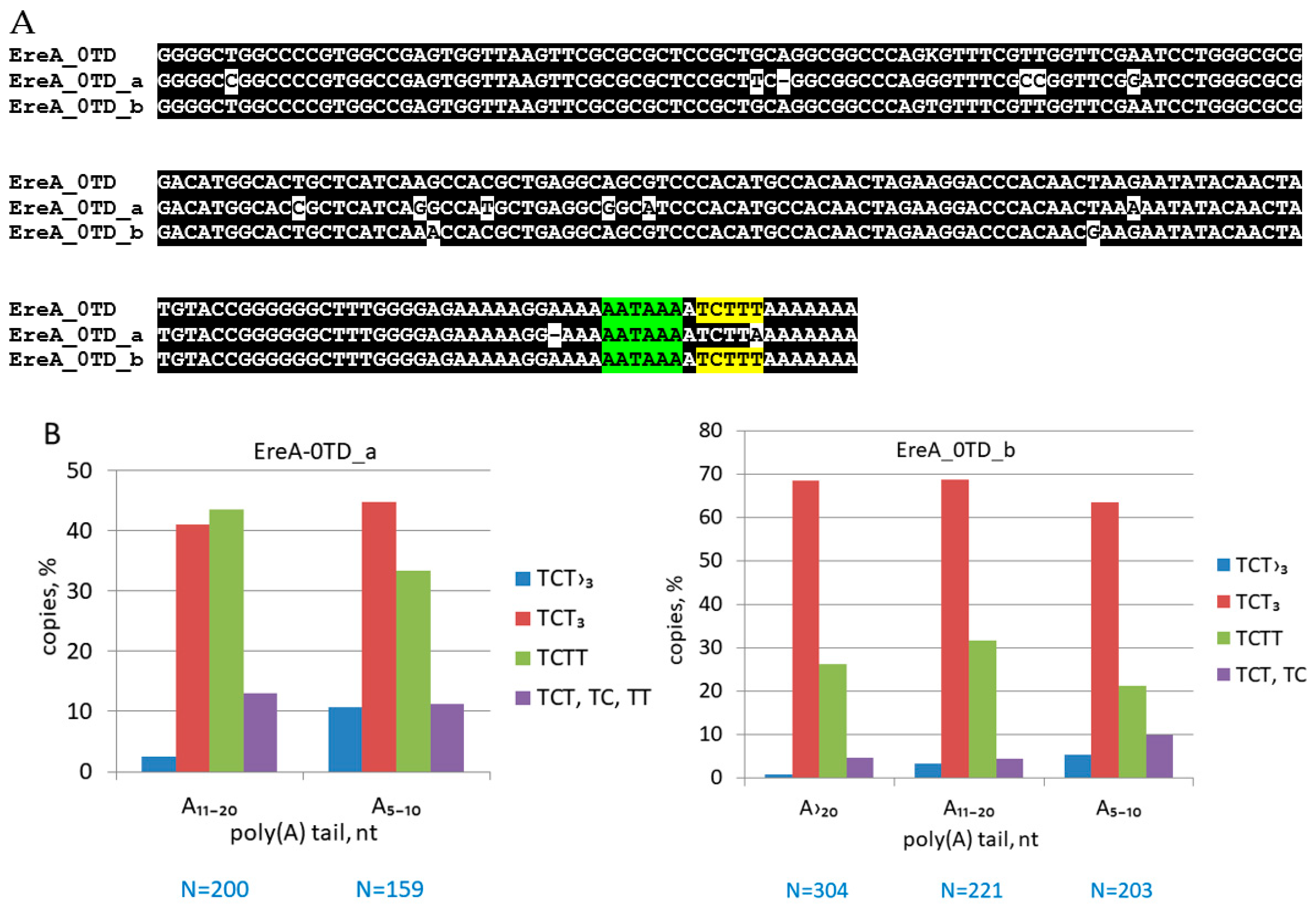
3.4. An Analysis of Young EreA_0TD Copies in the Horse Genome
3.5. Identification of β and τ Signals in EreA_0TD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kramerov, D.A.; Vassetzky, N.S. Short retroposons in eukaryotic genomes. Int. Rev. Cytol. 2005, 247, 165–221. [Google Scholar] [CrossRef]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef]
- Kramerov, D.A.; Vassetzky, N.S. SINEs. Wiley Interdiscip. Rev. RNA 2011, 2, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M.; Ferec, C.; Cooper, D.N. LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease: Mutation detection bias and multiple mechanisms of target gene disruption. J. Biomed. Biotechnol. 2006, 2006, 56182. [Google Scholar] [CrossRef]
- Kosushkin, S.A.; Ustyantsev, I.G.; Borodulina, O.R.; Vassetzky, N.S.; Kramerov, D.A. Tail Wags Dog’s SINE: Retropositional Mechanisms of Can SINE Depend on Its A-Tail Structure. Biology 2022, 11, 1403. [Google Scholar] [CrossRef]
- Ferrigno, O.; Virolle, T.; Djabari, Z.; Ortonne, J.P.; White, R.J.; Aberdam, D. Transposable B2 SINE elements can provide mobile RNA polymerase II promoters. Nat. Genet. 2001, 28, 77–81. [Google Scholar] [CrossRef]
- Su, M.; Han, D.; Boyd-Kirkup, J.; Yu, X.; Han, J.J. Evolution of Alu elements toward enhancers. Cell Rep. 2014, 7, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Policarpi, C.; Crepaldi, L.; Brookes, E.; Nitarska, J.; French, S.M.; Coatti, A.; Riccio, A. Enhancer SINEs Link Pol III to Pol II Transcription in Neurons. Cell Rep. 2017, 21, 2879–2894. [Google Scholar] [CrossRef]
- Krull, M.; Brosius, J.; Schmitz, J. Alu-SINE exonization: En route to protein-coding function. Mol. Biol. Evol. 2005, 22, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kirkness, E.F. Short interspersed elements (SINEs) are a major source of canine genomic diversity. Genome Res. 2005, 15, 1798–1808. [Google Scholar] [CrossRef]
- Krane, D.E.; Hardison, R.C. Short interspersed repeats in rabbit DNA can provide functional polyadenylation signals. Mol. Biol. Evol. 1990, 7, 1–8. [Google Scholar]
- Chen, C.; Ara, T.; Gautheret, D. Using Alu elements as polyadenylation sites: A case of retroposon exaptation. Mol. Biol. Evol. 2009, 26, 327–334. [Google Scholar] [CrossRef]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef]
- Sharma, H.; Valentine, M.N.Z.; Toki, N.; Sueki, H.N.; Gustincich, S.; Takahashi, H.; Carninci, P. Decryption of sequence, structure, and functional features of SINE repeat elements in SINEUP non-coding RNA-mediated post-transcriptional gene regulation. Nat. Commun. 2024, 15, 1400. [Google Scholar] [CrossRef] [PubMed]
- Ponicsan, S.L.; Kugel, J.F.; Goodrich, J.A. Genomic gems: SINE RNAs regulate mRNA production. Curr. Opin. Genet. Dev. 2010, 20, 149–155. [Google Scholar] [CrossRef]
- Zovoilis, A.; Cifuentes-Rojas, C.; Chu, H.P.; Hernandez, A.J.; Lee, J.T. Destabilization of B2 RNA by EZH2 Activates the Stress Response. Cell 2016, 167, 1788–1802.e13. [Google Scholar] [CrossRef]
- Borodulina, O.R.; Kramerov, D.A. Short interspersed elements (SINEs) from insectivores. Two classes of mammalian SINEs distinguished by A-rich tail structure. Mamm. Genome 2001, 12, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Vassetzky, N.S.; Kramerov, D.A. SINEBase: A database and tool for SINE analysis. Nucleic Acids Res. 2013, 41, D83–D89. [Google Scholar] [CrossRef]
- Ustyantsev, I.G.; Borodulina, O.R.; Kramerov, D.A. Identification of nucleotide sequences and some proteins involved in polyadenylation of RNA transcribed by Pol III from SINEs. RNA Biol. 2021, 18, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Borodulina, O.R.; Kramerov, D.A. Transcripts synthesized by RNA polymerase III can be polyadenylated in an AAUAAA-dependent manner. RNA 2008, 14, 1865–1873. [Google Scholar] [CrossRef]
- Borodulina, O.R.; Golubchikova, J.S.; Ustyantsev, I.G.; Kramerov, D.A. Polyadenylation of RNA transcribed from mammalian SINEs by RNA polymerase III: Complex requirements for nucleotide sequences. Biochim. Biophys. Acta 2016, 1859, 355–365. [Google Scholar] [CrossRef]
- Proudfoot, N.J. Ending the message: Poly(A) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef]
- Sun, Y.; Hamilton, K.; Tong, L. Recent molecular insights into canonical pre-mRNA 3′-end processing. Transcription 2020, 11, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, X.; Zhang, S.; Yuan, L.; Sun, Y. Molecular Insights into mRNA Polyadenylation and Deadenylation. Int. J. Mol. Sci. 2022, 23, 10985. [Google Scholar] [CrossRef] [PubMed]
- Boreikaite, V.; Passmore, L.A. 3′-End Processing of Eukaryotic mRNA: Machinery, Regulation, and Impact on Gene Expression. Annu. Rev. Biochem. 2023, 92, 199–225. [Google Scholar] [CrossRef] [PubMed]
- Borodulina, O.R.; Ustyantsev, I.G.; Kramerov, D.A. SINEs as Potential Expression Cassettes: Impact of Deletions and Insertions on Polyadenylation and Lifetime of B2 and Ves SINE Transcripts Generated by RNA Polymerase III. Int. J. Mol. Sci. 2023, 24, 14600. [Google Scholar] [CrossRef]
- Roy-Engel, A.M.; Salem, A.H.; Oyeniran, O.O.; Deininger, L.; Hedges, D.J.; Kilroy, G.E.; Batzer, M.A.; Deininger, P.L. Active Alu element “A-tails”: Size does matter. Genome Res. 2002, 12, 1333–1344. [Google Scholar] [CrossRef]
- Odom, G.L.; Robichaux, J.L.; Deininger, P.L. Predicting mammalian SINE subfamily activity from A-tail length. Mol. Biol. Evol. 2004, 21, 2140–2148. [Google Scholar] [CrossRef]
- Dewannieux, M.; Heidmann, T. Role of poly(A) tail length in Alu retrotransposition. Genomics 2005, 86, 378–381. [Google Scholar] [CrossRef]
- Roy-Engel, A.M. A tale of an A-tail: The lifeline of a SINE. Mob. Genet. Elem. 2012, 2, 282–286. [Google Scholar] [CrossRef]
- Batzer, M.A.; Deininger, P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, B.J.; Hedges, D.J.; Derbes, R.S.; Campos Sanchez, R.; Chiaromonte, F.; Makova, K.D.; Roy-Engel, A.M. Rescuing Alu: Recovery of new inserts shows LINE-1 preserves Alu activity through A-tail expansion. PLoS Genet. 2012, 8, e1002842. [Google Scholar] [CrossRef]
- Vassetzky, N.S.; Borodulina, O.R.; Ustyantsev, I.G.; Kosushkin, S.A.; Kramerov, D.A. Analysis of SINE Families B2, Dip, and Ves with Special Reference to Polyadenylation Signals and Transcription Terminators. Int. J. Mol. Sci. 2021, 22, 9897. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, M.; Ohshima, K.; Mukoyama, H.; Yasue, H.; Okada, N. A novel tRNA species as an origin of short interspersed repetitive elements (SINEs): Equine SINEs may have originated from tRNA(Ser). J. Mol. Biol. 1994, 239, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.C.; Lear, T.L.; Coogle, L.D.; Bailey, E. Two SINE families associated with equine microsatellite loci. Mamm. Genome 1999, 10, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Santagostino, M.; Khoriauli, L.; Gamba, R.; Bonuglia, M.; Klipstein, O.; Piras, F.M.; Vella, F.; Russo, A.; Badiale, C.; Mazzagatti, A.; et al. Genome-wide evolutionary and functional analysis of the Equine Repetitive Element 1: An insertion in the myostatin promoter affects gene expression. BMC Genet. 2015, 16, 126. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.D.; Tomii, K.; Katoh, K. Application of the MAFFT sequence alignment program to large data-reexamination of the usefulness of chained guide trees. Bioinformatics 2016, 32, 3246–3251. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef] [PubMed]
- MacHugh, D.E.; Larson, G.; Orlando, L. Taming the Past: Ancient DNA and the Study of Animal Domestication. Annu. Rev. Anim. Biosci. 2017, 5, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Der Sarkissian, C.; Ermini, L.; Schubert, M.; Yang, M.A.; Librado, P.; Fumagalli, M.; Jonsson, H.; Bar-Gal, G.K.; Albrechtsen, A.; Vieira, F.G.; et al. Evolutionary Genomics and Conservation of the Endangered Przewalski’s Horse. Curr. Biol. 2015, 25, 2577–2583. [Google Scholar] [CrossRef] [PubMed]
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I.; et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.; Westbury, M.V.; Russo, I.M.; Gopalakrishnan, S.; Rakotoarivelo, A.; Olsen, R.A.; Prost, S.; Tunstall, T.; Ryder, O.A.; Dalen, L.; et al. Interspecific Gene Flow and the Evolution of Specialization in Black and White Rhinoceros. Mol. Biol. Evol. 2020, 37, 3105–3117. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Westbury, M.V.; Dussex, N.; Mitchell, K.J.; Sinding, M.S.; Heintzman, P.D.; Duchene, D.A.; Kapp, J.D.; von Seth, J.; Heiniger, H.; et al. Ancient and modern genomes unravel the evolutionary history of the rhinoceros family. Cell 2021, 184, 4874–4885.e16. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, A.M.; Barraclough, T.G. The evolutionary reality of higher taxa in mammals. Proc. Biol. Sci. 2014, 281, 20132750. [Google Scholar] [CrossRef] [PubMed]
- Lim, Q.L.; Yong, C.S.Y.; Ng, W.L.; Ismail, A.; Rovie-Ryan, J.J.; Rosli, N.; Annavi, G. Genetic diversity and phylogenetic relationships of Malayan tapir (Tapirus indicus) populations in the Malay Peninsula based on mitochondrial DNA control region. Biodivers. Conserv. 2021, 30, 2433–2449. [Google Scholar] [CrossRef]
- Orioli, A.; Pascali, C.; Quartararo, J.; Diebel, K.W.; Praz, V.; Romascano, D.; Percudani, R.; van Dyk, L.F.; Hernandez, N.; Teichmann, M.; et al. Widespread occurrence of non-canonical transcription termination by human RNA polymerase III. Nucleic Acids Res. 2011, 39, 5499–5512. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Pu, Y.; Ma, Y.; Jiang, L. Whole-genome identification of transposable elements reveals the equine repetitive element insertion polymorphism in Chinese horses. Anim. Genet. 2023, 54, 144–154. [Google Scholar] [CrossRef]
- Secord, R.; Bloch, J.I.; Chester, S.G.; Boyer, D.M.; Wood, A.R.; Wing, S.L.; Kraus, M.J.; McInerney, F.A.; Krigbaum, J. Evolution of the earliest horses driven by climate change in the Paleocene-Eocene Thermal Maximum. Science 2012, 335, 959–962. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SINE Family | Domestic horse (−) | Przewalski horse (−) | Domestic donkey (−) | Plains zebra (−) | |
| Domestic horse (+) | EreA | 2241 | 12,592 | 11,496 | |
| EreB | 204 | 1974 | 600 | ||
| Przewalski horse (+) | EreA | 1301 | nd | 8961 | |
| EreB | 199 | nd | 552 | ||
| Domestic donkey (+) | EreA | 8491 | nd | nd | |
| EreB | 438 | nd | nd | ||
| Plains zebra (+) | EreA | 10,155 | 10,164 | nd | |
| EreB | 564 | 587 | nd |
| Black rhinoceros (−) | White rhinoceros (−) | Malayan tapir (−) | Lowland tapir (−) | |
| Black rhinoceros (+) | 25,373 EreA_0TD: 39% EreA_1 and 2TD: 4% EreB_2TD: 57% | |||
| White rhinoceros (+) | 30,285 EreA_0TD: 87% EreA_1 and 2TD: 2% EreB_2TD: 11% | |||
| Malayan tapir (+) | 25,754 EreB’_2TD: 100% | |||
| Lowland tapir (+) | 10,941 EreB’_2TD: 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ustyantsev, I.G.; Kosushkin, S.A.; Borodulina, O.R.; Vassetzky, N.S.; Kramerov, D.A. Ere, a Family of Short Interspersed Elements in the Genomes of Odd-Toed Ungulates (Perissodactyla). Animals 2024, 14, 1982. https://doi.org/10.3390/ani14131982
Ustyantsev IG, Kosushkin SA, Borodulina OR, Vassetzky NS, Kramerov DA. Ere, a Family of Short Interspersed Elements in the Genomes of Odd-Toed Ungulates (Perissodactyla). Animals. 2024; 14(13):1982. https://doi.org/10.3390/ani14131982
Chicago/Turabian StyleUstyantsev, Ilia G., Sergey A. Kosushkin, Olga R. Borodulina, Nikita S. Vassetzky, and Dmitri A. Kramerov. 2024. "Ere, a Family of Short Interspersed Elements in the Genomes of Odd-Toed Ungulates (Perissodactyla)" Animals 14, no. 13: 1982. https://doi.org/10.3390/ani14131982