

The Comparative Full-Length Genome Characterization of African Swine Fever Virus Detected in Thailand
 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Extraction
2.2. DNA Library Preparation and High-Throughput Sequencing
2.3. Characterization of TH1_22/CR
2.4. Retrieval of Public ASFV Genome Sequences and Alignment
2.5. Genetic Analysis
2.6. Analysis of ORFs for Sequence Diversity
2.7. Genetic Variation for SNPs and Single Indels
2.8. Amino Acid Variation for SNPs and Single Indels
2.9. Phylogenetic Tree of the Whole Genome
2.10. Intergenic Region Analysis
2.11. Tandem Repeat Sequence Variation
2.12. Oligonucleotide Indels
3. Results
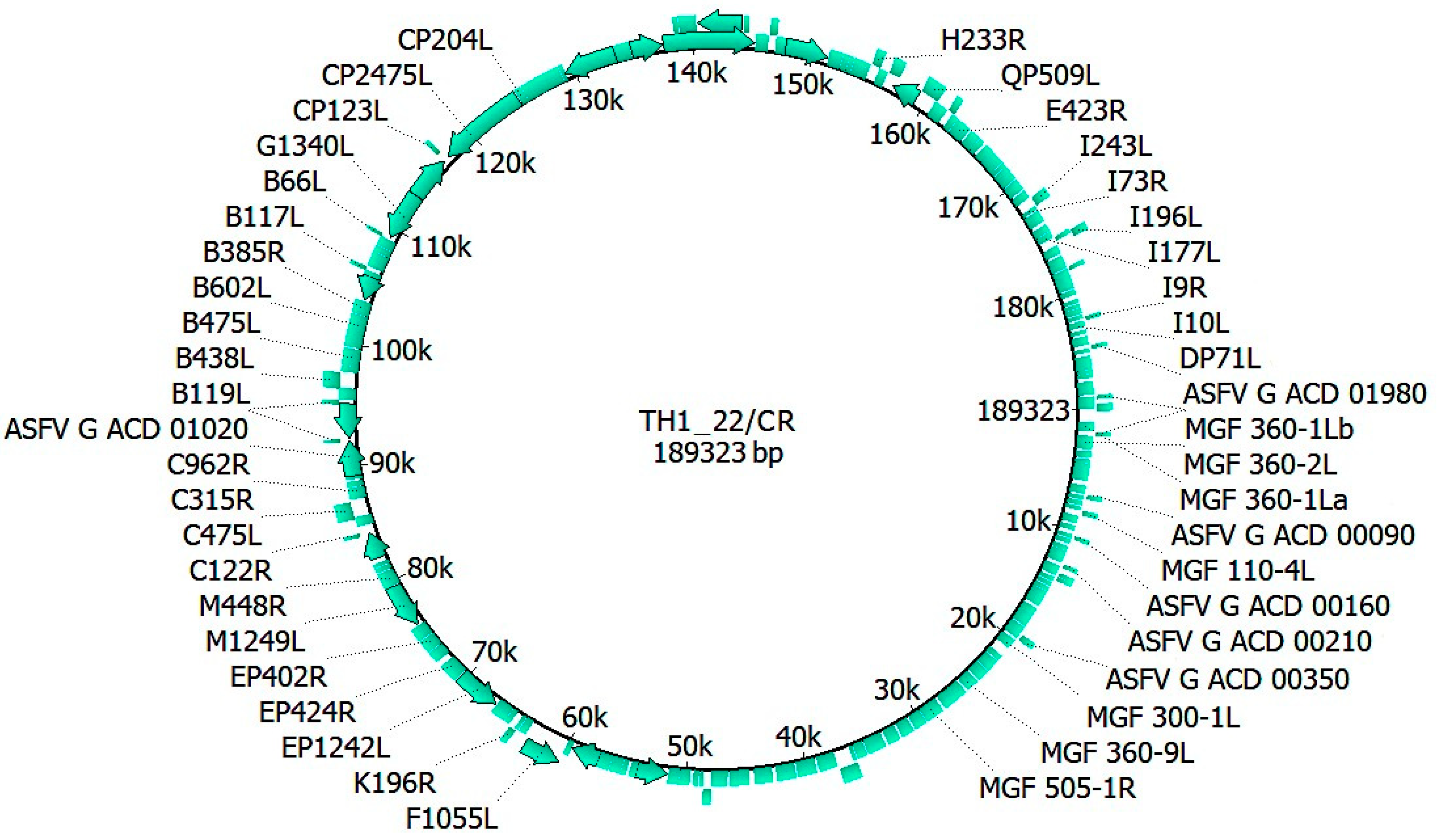
3.1. Whole Genome Sequence and Similarity Index of TH1_22/CR
3.2. Characterization of TH1_22/CR
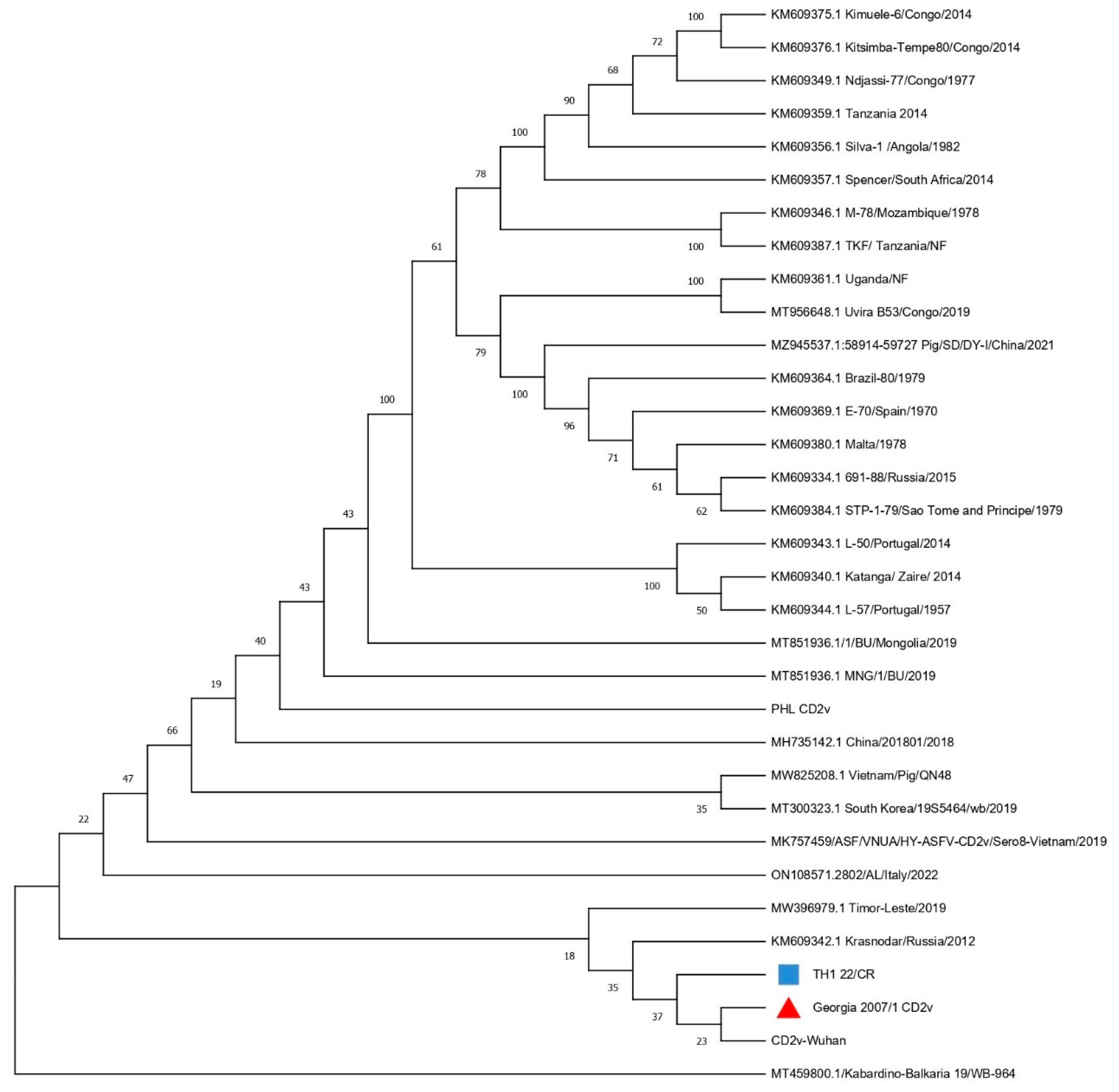
3.3. Phylogenetic Tree of the Whole Genome of TH1_22/CR
3.4. Comparative Analysis of the Variations in TH1_22/CR
3.5. Genetic Variations in the ORFs of TH1_22/CR
3.6. Amino Acid Variations in TH1_22/CR
3.7. Intergenic Variations in TH1_22/CR
Variation in TRS
3.8. Oligonucleotide Indels in TH1_22/CR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blome, S.; Franzke, K.; Beer, M. African swine fever–A review of current knowledge. Virus Res. 2020, 287, 198099. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Liu, R.; Zhang, X.; Li, F.; Wang, J.; Zhang, J.; Liu, X.; Wang, L.; Zhang, J.; Wu, X.; et al. Replication and virulence in pigs of the first African swine fever virus isolated in China. Emerg. Microbes Infect. 2019, 8, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.D.; Penrith, M.-L.; Cruciere, C.; Edrich, J.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef]
- Njau, E.P.; Machuka, E.M.; Cleaveland, S.; Shirima, G.M.; Kusiluka, L.J.; Okoth, E.A.; Pelle, R. African swine fever virus (ASFV): Biology, genomics and genotypes circulating in sub-Saharan Africa. Viruses 2021, 13, 2285. [Google Scholar] [CrossRef]
- Sauter-Louis, C.; Conraths, F.J.; Probst, C.; Blohm, U.; Schulz, K.; Sehl, J.; Fischer, M.; Forth, J.H.; Zani, L.; Depner, K. African swine fever in wild boar in Europe—A review. Viruses 2021, 13, 1717. [Google Scholar] [CrossRef]
- Xin, G.; Kuang, Q.; Le, S.; Wu, W.; Gao, Q.; Gao, H.; Xu, Z.; Zheng, Z.; Lu, G.; Gong, L. Origin, genomic diversity and evolution of African swine fever virus in East Asia. Virus Evol. 2023, 9, vead060. [Google Scholar] [CrossRef]
- Giammarioli, M.; Marcacci, M.; Scicluna, M.T.; Cersini, A.; Torresi, C.; Curini, V.; Ancora, M.; Rinaldi, A.; Sala, M.G.; Rossi, E.; et al. Complete Genome of African Swine Fever Virus Genotype II in Central Italy. Microbiol. Resour. Announc. 2023, 12, e0136422. [Google Scholar] [CrossRef]
- Bao, J.; Zhang, Y.; Shi, C.; Wang, Q.; Wang, S.; Wu, X.; Cao, S.; Xu, F.; Wang, Z. Genome-wide diversity analysis of African swine fever virus based on a curated dataset. Animals 2022, 12, 2446. [Google Scholar] [CrossRef]
- Mazloum, A.; van Schalkwyk, A.; Shotin, A.; Zinyakov, N.; Igolkin, A.; Chernishev, R.; Debeljak, Z.; Korennoy, F.; Sprygin, A.V. Whole-genome sequencing of African swine fever virus from wild boars in the Kaliningrad region reveals unique and distinguishing genomic mutations. Front. Vet. Sci. 2022, 9, 1019808. [Google Scholar] [CrossRef]
- Mazloum, A.; van Schalkwyk, A.; Chernyshev, R.; Igolkin, A.; Heath, L.; Sprygin, A. A Guide to Molecular Characterization of Genotype II African Swine Fever Virus: Essential and Alternative Genome Markers. Microorganisms 2023, 11, 642. [Google Scholar] [CrossRef]
- Montgomery, R.E. On a form of swine fever occurring in British East Africa (Kenya Colony). J. Comp. Pathol. Ther. 1921, 34, 159–191. [Google Scholar] [CrossRef]
- Couacy-Hymann, E. African swine fever in sub-Saharan African countries. In Transboundary Animal Diseases in Sahelian Africa and Connected Regions; Springer: Cham, Switzerland, 2019; pp. 323–344. [Google Scholar]
- Fauquet, C.M.; Mayo, M.A.; Maniloff, J.; Desselberger, U.; Ball, L.A. Virus Taxonomy: VIIIth Report of the International Committee on Taxonomy of Viruses; Academic Press: Cambridge, MA, USA, 2005. [Google Scholar]
- Wu, K.; Liu, J.; Wang, L.; Fan, S.; Li, Z.; Li, Y.; Yi, L.; Ding, H.; Zhao, M.; Chen, J. Current state of global African swine fever vaccine development under the prevalence and transmission of ASF in China. Vaccines 2020, 8, 531. [Google Scholar] [CrossRef] [PubMed]
- Guinat, C.; Gogin, A.; Blome, S.; Keil, G.; Pollin, R.; Pfeiffer, D.U.; Dixon, L. Transmission routes of African swine fever virus to domestic pigs: Current knowledge and future research directions. Vet. Rec. 2016, 178, 262–267. [Google Scholar] [CrossRef]
- Lu, G.; Pan, J.; Zhang, G. African swine fever virus in Asia: Its rapid spread and potential threat to unaffected countries. J. Infect. 2020, 80, 350–371. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Zhang, J.Y.; Yang, J.J.; Yang, J.M.; Han, X.; Mi, L.J. Identification of a natural variant of African swine fever virus in China. Chin. J. Vet. Sci. 2023, 41, 199–207. [Google Scholar]
- Kim, G.; Park, J.E.; Kim, S.J.; Kim, Y.; Kim, W.; Kim, Y.K.; Jheong, W. Complete genome analysis of the African swine fever virus isolated from a wild boar responsible for the first viral outbreak in Korea, 2019. Front. Vet. Sci. 2022, 9, 1080397. [Google Scholar] [CrossRef]
- Mighell, E.; Ward, M.P. African Swine Fever spread across Asia, 2018–2019. Transbound. Emerg. Dis. 2021, 68, 2722–2732. [Google Scholar] [CrossRef]
- Senthilkumar, D.; Rajukumar, K.; Venkatesh, G.; Singh, F.; Tosh, C.; Kombiah, S.; Dubey, C.K.; Chakravarty, A.; Barman, N.N.; Singh, V.P. Complete genome analysis of African swine fever virus isolated from domestic pigs during the first ASF outbreaks in India. Transbound. Emerg. Dis. 2022, 69, e2020–e2027. [Google Scholar] [CrossRef]
- Le, V.P.; Ahn, M.J.; Kim, J.S.; Jung, M.C.; Yoon, S.W.; Trinh, T.B.N.; Le, T.N.; Kim, H.K.; Kang, J.A.; Lim, J.W.; et al. A Whole-Genome Analysis of the African Swine Fever Virus That Circulated during the First Outbreak in Vietnam in 2019 and Subsequently in 2022. Viruses 2023, 15, 1945. [Google Scholar] [CrossRef]
- World Organization for Animal Health (WOAH). African Swine Fever Situation Report. Available online: https://wahis.woah.org/#/in-review/4236?reportId=158536&frompage=event-dashboard-url (accessed on 10 January 2023).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 3 March 2024).
- Kim, H.J.; Cho, K.H.; Ryu, J.H.; Jang, M.K.; Chae, H.G.; Choi, J.D.; Nah, J.J.; Kim, Y.J.; Kang, H.E. Isolation and Genetic Characterization of African Swine Fever Virus from Domestic Pig Farms in South Korea, 2019. Viruses 2020, 12, 1237. [Google Scholar] [CrossRef]
- Lubisi, B.A.; Bastos, A.; Dwarka, R.; Vosloo, W. Molecular epidemiology of African swine fever in East Africa. Arch. Virol. 2005, 150, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- Mulumba-Mfumu, L.K.; Saegerman, C.; Dixon, L.K.; Madimba, K.C.; Kazadi, E.; Mukalakata, N.T.; Oura, C.A.; Chenais, E.; Masembe, C.; Ståhl, K. African swine fever: Update on Eastern, Central and Southern Africa. Transbound. Emerg. Dis. 2019, 66, 1462–1480. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, M.C.; Reoyo, A.d.l.T.; Fernández-Pinero, J.; Iglesias, I.; Muñoz, M.J.; Arias, M.L. African swine fever: A global view of the current challenge. Porc. Health Manag. 2015, 1, 1–14. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Dharmayanti, N.I.; Sendow, I.; Ratnawati, A.; Settypalli, T.B.K.; Saepulloh, M.; Dundon, W.G.; Nuradji, H.; Naletoski, I.; Cattoli, G.; Lamien, C.E. African swine fever in North Sumatra and West Java provinces in 2019 and 2020, Indonesia. Transbound. Emerg. Dis. 2021, 68, 2890–2896. [Google Scholar] [CrossRef] [PubMed]
- Alkhamis, M.A.; Gallardo, C.; Jurado, C.; Soler, A.; Arias, M.; Sanchez-Vizcaino, J.M. Phylodynamics and evolutionary epidemiology of African swine fever p72-CVR genes in Eurasia and Africa. PLoS ONE 2018, 13, e0192565. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Hakizimana, J.N.; Ntirandekura, J.B.; Yona, C.; Nyabongo, L.; Kamwendo, G.; Chulu, J.L.; Ntakirutimana, D.; Kamana, O.; Nauwynck, H.; Misinzo, G. Complete genome analysis of African swine fever virus responsible for outbreaks in domestic pigs in 2018 in Burundi and 2019 in Malawi. Trop. Anim. Health Prod. 2021, 53, 438. [Google Scholar] [CrossRef]
- Katoh, K.; Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 2010, 26, 1899–1900. [Google Scholar] [CrossRef]
- Ebwanga, E.J.; Ghogomu, S.M.; Paeshuyse, J. Molecular characterization of ASFV and differential diagnosis of erysipelothrix in ASFV-infected pigs in pig production regions in cameroon. Vet. Sci. 2022, 9, 440. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, J.Y.; Tseren-Ochir, E.O.; Lee, D.H.; Nahm, S.S.; Gladue, D.P.; Borca, M.V.; Song, C.S.; Risatti, G.R. Whole genome sequencing and phylogenetic analysis of African swine fever virus detected in a backyard pig in Mongolia, 2019. Front. Vet. Sci. 2023, 10, 1094052. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Ge, S.; Zhang, Y.; Wu, X.; Wang, Z. A systematic review of genotypes and serogroups of African swine fever virus. Virus Genes 2022, 58, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Casado, N.; Soler, A.; Djadjovski, I.; Krivko, L.; Madueño, E.; Nieto, R.; Perez, C.; Simon, A.; Ivanova, E. A multi gene-approach genotyping method identifies 24 genetic clusters within the genotype II-European African swine fever viruses circulating from 2007 to 2022. Front. Vet. Sci. 2023, 10, 1112850. [Google Scholar] [CrossRef]
- Ito, S.; Kawaguchi, N.; Bosch, J.; Aguilar-Vega, C.; Sánchez-Vizcaíno, J.M. What can we learn from the five-year African swine fever epidemic in Asia? Front. Vet. Sci. 2023, 10, 1273417. [Google Scholar] [CrossRef]
- Granja, A.G.; Nogal, M.L.; Hurtado, C.; Vila, V.; Carrascosa, A.L.; Salas, M.L.; Fresno, M.; Revilla, Y. The viral protein A238L inhibits cyclooxygenase-2 expression through a nuclear factor of activated T cell-dependent transactivation pathway. J. Biol. Chem. 2004, 279, 53736–53746. [Google Scholar] [CrossRef]
- Granja, A.G.; Nogal, M.L.; Hurtado, C.; Del Aguila, C.; Carrascosa, A.L.; Salas, M.L.; Fresno, M.; Revilla, Y. The viral protein A238L inhibits TNF-α expression through a CBP/p300 transcriptional coactivators pathway. J. Immunol. 2006, 176, 451–462. [Google Scholar] [CrossRef]
- Ramirez-Medina, E.; Vuono, E.; Pruitt, S.; Rai, A.; Silva, E.; Espinoza, N.; Zhu, J.; Velazquez-Salinas, L.; Borca, M.V.; Gladue, D.P. Development and in vivo evaluation of a MGF110-1L deletion mutant in African swine fever strain Georgia. Viruses 2021, 13, 286. [Google Scholar] [CrossRef]
- Li, D.; Peng, J.; Wu, J.; Yi, J.; Wu, P.; Qi, X.; Ren, J.; Peng, G.; Duan, X.; Ru, Y. African swine fever virus MGF-360-10L is a novel and crucial virulence factor that mediates ubiquitination and degradation of JAK1 by recruiting the E3 ubiquitin ligase HERC5. Mbio 2023, 14, e00606–e00623. [Google Scholar] [CrossRef]
- Yang, K.; Xue, Y.; Niu, T.; Li, X.; Cheng, M.; Bao, M.; Zou, B.; Shi, C.; Wang, J.; Yang, W. African swine fever virus MGF505-7R protein interacted with IRF7and TBK1 to inhibit type I interferon production. Virus Res. 2022, 322, 198931. [Google Scholar] [CrossRef]
- Ran, Y.; Li, D.; Xiong, M.-G.; Liu, H.-N.; Feng, T.; Shi, Z.-W.; Li, Y.-H.; Wu, H.-N.; Wang, S.-Y.; Zheng, H.-X. African swine fever virus I267L acts as an important virulence factor by inhibiting RNA polymerase III-RIG-I-mediated innate immunity. PLoS Pathog. 2022, 18, e1010270. [Google Scholar] [CrossRef]
- Wen, Y.; Duan, X.; Ren, J.; Zhang, J.; Guan, G.; Ru, Y.; Li, D.; Zheng, H. African Swine Fever Virus I267L Is a Hemorrhage-Related Gene Based on Transcriptome Analysis. Microorganisms 2024, 12, 400. [Google Scholar] [CrossRef]
- Wang, G.; Xie, M.; Wu, W.; Chen, Z. Structures and functional diversities of ASFV proteins. Viruses 2021, 13, 2124. [Google Scholar] [CrossRef] [PubMed]
- Bisimwa, P.N.; Ongus, J.R.; Tiambo, C.K.; Machuka, E.M.; Bisimwa, E.B.; Steinaa, L.; Pelle, R. First detection of African swine fever (ASF) virus genotype X and serogroup 7 in symptomatic pigs in the Democratic Republic of Congo. Virol. J. 2020, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pavone, S.; Iscaro, C.; Dettori, A.; Feliziani, F. African swine fever: The state of the art in Italy. Animals 2023, 13, 2998. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Accession No. | Isolate Name | City | Country | Genotype | Year | Genome | Length (bp) |
|---|---|---|---|---|---|---|---|---|
| 1 | FR682468 | Georgia 2007/1 | Georgia | Georgia | II | 2007 | Full-length | 190,584 |
| 2 | MK333180 | HLJ/2018 | Harbin, Heilongjiang | China | II | 2018 | Full-length | 189,404 |
| 3 | MN393476 | Wuhan 2019-1 | Wuhan, Hubei | China | II | 2019 | Full-length | 190,576 |
| 4 | MK128995 | Anhui/XCGQ | Qingdao, Shandong | China | II | 2018 | Full-length | 189,393 |
| 5 | OP856591 | LN/2018/1 | Qingdao, Shandong | China | II | 2018 | Full-length | 189,397 |
| 6 | MT496893 | GZ201801 | Guangzhou, Guangdong | China | II | 2018 | Full-length | 189,393 |
| 7 | MK645909 | wbBS01 | Changchun, Jilin | China | II | 2018 | Full-length | 189,394 |
| 8 | MZ614662 | CADC HN09 | Beijing | China | II | 2019 | Full-length | 190,257 |
| 9 | MN172368 | CAS19-01 | Wuhan, Hubei | China | II | 2019 | Full-length | 189,405 |
| 10 | MW033528 | wbShX01 | Changchun, Jilin | China | II | 2019 | Full-length | 189,401 |
| 11 | MK940252 | InnerMongolia-AES01 | Changchun, Jilin | China | II | 2019 | Full-length | 189,403 |
| 12 | OM161110 | SY-1 | Wuhan, Hubei | China | II | 2020 | Full-length | 189,404 |
| 13 | MW521382 | HuB20 | Changchun, Jilin | China | II | 2020 | Full-length | 188,643 |
| 14 | MW656282 | Heilongjiang/HRB1/2020 | Harbin, Heilongjiang | China | II | 2020 | Full-length | 189,355 |
| 15 | OL310288 | China/2020 | Lanzhou, Gansu | China | II | 2020 | Full-length | 188,072 |
| 16 | OP612151 | SY-2 | Wuhan, Hubei | China | II | 2021 | Full-length | 189,404 |
| 17 | ON456300 | Yangzhou | Yangzhou | China | II | 2021 | Full-length | 187,951 |
| 18 | MT882025 | VN/QP-ASFV1(2019) | Hanoi | Vietnam | II | 2019 | Full-length | 189,081 |
| 19 | ON402789 | VN/2021 | Ninh Kieu District, Can Tho | Vietnam | II | 2021 | Full-length | 189,487 |
| 20 | MW049116 | Yeoncheon1/2019 | Gimcheon, Gyeongsangbuk-do | Korea | II | 2019 | Full-length | 187,848 |
| 21 | OR159219 | S-S-VR-413000-00002 | Ik-San, Jeollabuk-Do | Korea | II | 2019 | Full-length | 189,400 |
| 22 | OR159218 | S-S-VR-413000-00008 | Ik-San, Jeollabuk-Do | Korea | II | 2019 | Full-length | 189,429 |
| 23 | MT748042 | PaJu1/2019 | Gimcheon, Gyeongsangbuk-do | Korea | II | 2019 | Full-length | 187,848 |
| 24 | OR159217 | S-S-VR-413000-00015 | Ik-San, Jeollabuk-Do | Korea | II | 2019 | Full-length | 189,411 |
| 25 | MW791752 | ASFV2020-008-B | Manila | Philippines | II | 2020 | Full-length | 190,565 |
| 26 | MW791759 | ASFV2020-021-B | Manila | Philippines | II | 2020 | Full-length | 190,571 |
| 27 | MW791760 | ASFV2019-003-B | Manila | Philippines | II | 2019 | Full-length | 190,571 |
| 28 | MW791761 | ASFV2020-003-B | Manila | Philippines | II | 2020 | Full-length | 190,571 |
| 29 | ON963982 | PHL/Strain A4 | Los Banos, Laguna | Philippines | II | 2021 | Full-length | 192,265 |
| 30 | MW396979 | Timor-Leste/2019/1 | Timor-Leste | Timor-Leste | II | 2019 | Full-length | 192,237 |
| 31 | TH1_CR/2022 | Chiang Rai | Thailand | II | 2022 | Full-length | 189,395 |
| Sequence Identity Matrix | TH1_22/CR_Thailand | FR682468.2 Georgia 2007/1_Georgia | MN393476_Wuhan/2019-1_China | MK333180_HLJ_China/2018 | MT882025.1 VN/QP-ASFV1(2019)_Vietnam | ON402789_Serotype 8 Genotype 2 (VN/2021)_Vietnam | MW791760_2019-003-B_2019/Philippines | ON963982_PHL/StrainA4_Philippines (2021) | MW049116_Pig/Yeoncheon1/2019_Korea (2019) | OR159218_S-S-VR-413000-00008_Korea (2019) |
|---|---|---|---|---|---|---|---|---|---|---|
| TH1_22/CR_Thailand | ID | 99.9 | 99.9 | 99.9 | 99.7 | 99.2 | 99.9 | 99.9 | 99.1 | 99.9 |
| FR682468.2 Georgia 2007/1_Georgia | 99.9 | ID | 99.9 | 99.9 | 99.7 | 99.2 | 99.9 | 99.9 | 99.1 | 99.9 |
| MN393476_Wuhan/2019-1_China | 99.9 | 99.9 | ID | 99.9 | 99.7 | 99.2 | 99.9 | 99.9 | 99.1 | 99.9 |
| MK333180_HLJ_China/2018 | 99.9 | 99.9 | 99.9 | ID | 99.7 | 99.2 | 99.9 | 99.9 | 99.1 | 99.9 |
| MT882025.1 VN/QP-ASFV1(2019) Vietnam | 99.6 | 99.6 | 99.6 | 99.6 | ID | 99 | 99.7 | 99.7 | 98.9 | 99.7 |
| ON402789_Serotype 8 genotype 2 (VN/2021) Vietnam | 99.2 | 99.2 | 99.2 | 99.2 | 98.9 | ID | 99.2 | 99.2 | 99.6 | 99.2 |
| MW791760_2019-003-B_2019/Philippines | 99.9 | 99.9 | 99.9 | 99.9 | 99.6 | 99.2 | ID | 99.9 | 99.1 | 99.9 |
| ON963982_PHL/StrainA4_Philippines (2021) | 99.8 | 99.8 | 99.9 | 99.9 | 99.6 | 99.1 | 99.9 | ID | 99.1 | 99.9 |
| MW049116_pig/Yeoncheon1/2019_Korea (2019) | 99.1 | 99.1 | 99.1 | 99.1 | 98.8 | 99.6 | 99.1 | 99 | ID | 99.1 |
| OR159218_S-S-VR-413000-00008_Korea (2019) | 99.8 | 99.9 | 99.9 | 99.9 | 99.6 | 99.2 | 99.9 | 99.8 | 99.1 | ID |
| Regions | Total Genes in Region | No. of Variable Genes | Percentage | Genes Variable in Asia |
|---|---|---|---|---|
| Region-I (From 1 to 47,170 bp) | 59 | 11 | 18.64 | MGF 360-2L MGF 110-1L MGF 110-3L MGF 110-7L MGF 360-4L MGF 360-10L MGF 300-2R MGF 505-1R MGF 360-14L MGF 505-4R MGF 505-9R |
| Region-II (From 47,171 to 101,665 bp) | 49 | 7 | 14.2 | A238L EP364R F165R EP153R M1249L C84L C717R |
| Region-III (From 101,666 to 162,619 bp) | 30 | 5 | 16.66 | G1211R O174L NP1450L NP419L D1133L |
| Region-IV (From 162,620 to 190,169 bp) | 57 | 5 | 8.77 | I267L MGF-360 16R I10L ASFV G ACD 1980 DP60R |
| Total | 195 | 28 | 14.35 |
| Gene Name | MGF 360-2L | MGF 110-1L | MGF 110-3L | MGF 110-7L | MGF 360-4L | MGF 360-10L | MGF 300-2R | MGF 505-1R | MGF 360-14L | MGF 505-4R | MGF 505-9R | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Regions | Region I | |||||||||||||||||
| Location in Gene | 365 | 51 | 42 | 235 | 236 | 249 | 328 | 365 | 438 | 591 | 53 | 811 | 216 | 1155 | 224 | 250 | 1020 | 962 |
| FR682468.2 Georgia 2007/1_Georgia | A | C | G | G | T | A | A | C | C | C | T | G | A | G | X | T | T | A |
| TH1_22/CR Thailand (2022) | A | T | G | G | T | A | A | C | C | T | C | G | A | G | X | T | T | G |
| MN393476 Wuhan/2019-1_China | A | T | G | G | T | A | A | C | C | C | C | G | A | G | C | T | T | G |
| MK333180 HLJ_China/2018 | A | T | G | G | T | A | A | C | C | C | C | G | A | G | X | T | T | G |
| MT882025.1 VN/QP-ASFV1(2019)_Vietnam | A | T | G | C | C | A | A | C | C | C | C | G | A | G | C | T | T | G |
| ON402789_Serotype 8 genotype 2 (VN/2021)_Vietnam | X | T | A | C | C | G | C | X | T | C | C | G | A | G | X | X | T | G |
| MW791760 2019-003-B_2019/Philippines | X | T | G | G | T | A | A | X | C | C | C | G | A | G | X | T | T | G |
| ON963982 PHL/StrainA4_Philippines(2021) | X | T | G | G | T | A | A | X | C | C | C | G | X | X | X | T | X | G |
| MW049116 pig/Yeoncheon1/2019_Korea(2019) | A | T | G | G | T | A | A | C | C | C | C | C | A | G | X | T | T | G |
| OR159218 S-S-VR-413000-00008_Korea(2019) | A | T | G | G | T | A | A | C | C | C | C | C | A | G | X | T | T | G |
| Gene Name | A238L | EP364R | F165R | EP153R | M1249L | C84L | C717R | G1211R | O174L | NP1450L | NP419L | D1133L | ||||||
| Regions | Region II | Region III | ||||||||||||||||
| Location in Gene | 297 | 791 | 69 | 52 | 3461 | 156 | 797 | 3296 | 128 | 926 | 20 | 935 | 278 | 1366 | 3241 | |||
| FR682468.2 Georgia 2007/1_Georgia | C | T | T | T | T | T | T | A | T | T | C | C | T | G | C | |||
| TH1_22/CR Thailand (2022) | T | T | T | T | T | T | T | A | T | T | T | C | T | G | C | |||
| MN393476 Wuhan/2019-1_China | C | T | T | T | T | T | G | A | T | T | T | C | T | G | C | |||
| MK333180 HLJ_China/2018 | C | T | T | T | T | T | T | A | T | T | T | C | X | G | C | |||
| MT882025.1 VN/QP-ASFV1(2019)_Vietnam | C | T | T | T | T | T | T | A | T | T | T | T | T | G | C | |||
| ON402789_Serotype 8 genotype 2 (VN/2021)_Vietnam | C | T | T | T | T | T | T | A | T | T | T | C | T | G | C | |||
| MW791760 2019-003-B_2019/Philippines | C | T | T | T | T | T | T | A | T | T | T | C | T | G | C | |||
| ON963982 PHL/StrainA4_Philippines(2021) | C | T | X | X | X | X | T | X | X | X | T | C | T | X | X | |||
| MW049116 pig/Yeoncheon1/2019_Korea(2019) | C | X | T | T | T | T | T | A | T | T | T | C | T | G | C | |||
| OR159218 S-S-VR-413000-00008_Korea(2019) | C | T | T | T | T | T | T | A | T | T | T | C | T | G | C | |||
| Gene Name | I267L | MGF 360-16R | I10L | ASFV G ACD 01980 | DP60R | NCR | NCR | NCR | NCR | |||||||||
| Regions | Region IV | |||||||||||||||||
| Location in Gene | 222 | 260 | 898 | 415 | 179 | 104 | 189,406 | 189,433 | 189,439 | 189,459 | ||||||||
| FR682468.2 Georgia 2007/1_Georgia | T | G | A | T | T | X | A | C | G | T | ||||||||
| TH1_22/CR Thailand (2022) | A | G | A | T | T | A | G | G | A | A | ||||||||
| MN393476 Wuhan/2019-1_China | A | G | A | T | T | A | A | C | G | T | ||||||||
| MK333180 HLJ_China/2018 | A | G | A | T | T | A | A | C | G | T | ||||||||
| MT882025.1 VN/QP-ASFV1(2019)_Vietnam | A | G | A | T | T | A | A | C | G | T | ||||||||
| ON402789_Serotype 8 genotype 2 (VN/2021)_Vietnam | A | G | A | T | T | A | A | C | G | T | ||||||||
| MW791760 2019-003-B_2019/Philippines | A | G | A | T | T | A | A | C | G | T | ||||||||
| ON963982 PHL/StrainA4_Philippines(2021) | A | X | X | X | X | X | A | C | G | T | ||||||||
| MW049116 pig/Yeoncheon1/2019_Korea(2019) | A | G | A | T | T | A | A | C | G | T | ||||||||
| OR159218 S-S-VR-413000-00008_Korea(2019) | A | G | A | T | T | A | A | C | G | T | ||||||||
| Gene Name | MGF 360-2L | MGF 110-1L | MGF 110-3L | MGF 110-7L | MGF 360-4L | MGF 360-10L | MGF 300-2R | MGF 505-1R | MGF 360-14L | MGF 505-4R | MGF 505-9R | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Regions | Region I | |||||||||||||||
| Location in Gene | 242 | 19 | 18 | 47 | 112 | 13 | 192 | 243 | 18 | 271 | 72 | 282 | 276 | 184 | 340 | 323 |
| FR682468.2 Georgia 2007/1_Georgia | Leu | Pro | Leu | Asp | His | Ala | Ala | Val | Val | Glu | Ile | Ala | Met | X | Leu | Lys |
| TH1_22/CR Thailand (2022) | Leu | Leu | Leu | Asp | His | Ala | Thr | Val | Ala | Glu | Ile | Ala | Met | X | Leu | Glu |
| MN393476 Wuhan/2019-1_China | Leu | Leu | Leu | Asp | His | Ala | Ala | Val | Ala | Glu | Ile | Ala | Met | Met | Leu | Glu |
| MK333180 HLJ_China/2018 | Leu | Leu | Leu | Asp | His | Ala | Ala | Val | Ala | Glu | Ile | Ala | Met | X | Leu | Glu |
| MT882025.1 VN/QP-ASFV1(2019)Vietnam | Leu | Leu | Leu | Asp | His | Ala | Ala | Val | Ala | Glu | Ile | Ala | Met | Met | Leu | Glu |
| ON402789 VN/2021_Vietnam | X | Leu | Val | Arg | Tyr | X | Ala | Ile | Ala | Glu | Ile | Ala | X | X | Leu | Glu |
| MW791760 2019-003-B_2019/Philippines | X | Leu | Leu | Asp | His | X | Ala | Val | Ala | Glu | Ile | Ala | Met | X | Leu | Glu |
| ON963982 PHL/StrainA4_Philippines(2021) | X | Leu | Leu | Asp | His | X | Ala | Val | Ala | Glu | X | X | Met | X | X | Glu |
| MW049116 pig/Yeoncheon1/2019_Korea(2019) | Leu | Leu | Leu | Asp | His | Ala | Ala | Val | Ala | Glu | Ile | Ala | Met | X | Leu | Glu |
| OR159218 S-S-VR-413000-00008_Korea(2019) | Leu | Leu | Leu | Asp | His | Ala | Ala | Val | Ala | Glu | Ile | Ale | Met | X | Leu | Glu |
| Gene Name | A238L | EP364R | F165R | EP153R | M1249L | C84L | C717R | G1211R | O174L | NP419L | D1133L | |||||
| Regions | Region II | Region III | ||||||||||||||
| Location in Gene | 132 | 264 | 23 | 18 | 92 | 26 | 266 | 1099 | 133 | 109 | 414 | 54 | 679 | 1042 | ||
| FR682468.2 Georgia 2007/1_Georgia | Ala | Leu | Asn | Lys | Lys | Arg | Val | Glu | Lys | Arg | Asn | Gly | Pro | Gln | ||
| TH1_22/CR Thailand (2022) | Thr | Leu | Asn | Lys | Lys | Arg | Val | Glu | Lys | Arg | Ser | Gly | Pro | Gln | ||
| MN393476 Wuhan/2019-1_China | Ala | Leu | Asn | Lys | Lys | Arg | Glu | Glu | Lys | Arg | Ser | Gly | Pro | Gln | ||
| MK333180 HLJ_China/2018 | Ala | Leu | Asn | Lys | Lys | Arg | Val | Glu | Lys | Arg | Ser | Gly | Pro | X | ||
| MT882025.1 VN/QP-ASFV1(2019)Vietnam | Ala | Leu | Asn | Lys | Lys | Arg | Val | Glu | Lys | His | Ser | Gly | Pro | Gln | ||
| ON402789 VN/2021_Vietnam | Ala | Leu | Asn | Lys | Lys | Arg | Val | Glu | Lys | Arg | Ser | Gly | Pro | Gln | ||
| MW791760 2019-003-B_2019/Philippines | Ala | Leu | Asn | Lys | Lys | Arg | Val | Glu | Lys | Arg | Ser | Gly | Pro | Gln | ||
| ON963982 PHL/StrainA4_Philippines(2021) | Ala | X | X | X | X | X | Val | X | X | Arg | Ser | X | X | Gln | ||
| MW049116 pig/Yeoncheon1/2019_Korea(2019) | Ala | Leu | Asn | Lys | Lys | Arg | Val | Glu | Lys | Arg | Ser | Gly | Pro | Gln | ||
| OR159218 S-S-VR-413000-00008_Korea(2019) | Ala | Leu | Asn | Lys | Lys | Arg | Val | Glu | Lys | Arg | Ser | Gly | Pro | Gln | ||
| Gene Name | I267L | MGF 360-16R | I10L | ASFV G ACD 01980 | DP60R | |||||||||||
| Regions | Region IV | |||||||||||||||
| Location in Gene | 195 | 87 | 300 | 91 | 60 | 238 | 35 | |||||||||
| FR682468.2 Georgia 2007/1_Georgia | Ile | Trp | Lys | Asn | Ile | Lys | X | |||||||||
| TH1_22/CR Thailand (2022) | Phe | Trp | Lys | Asn | Ile | Lys | Gln | |||||||||
| MN393476 Wuhan/2019-1_China | Phe | Trp | Lys | Asn | Ile | Lys | Gln | |||||||||
| MK333180 HLJ_China/2018 | Phe | Trp | Lys | Asn | Ile | Lys | Gln | |||||||||
| MT882025.1 VN/QP-ASFV1(2019)Vietnam | Phe | Trp | Lys | Asn | Ile | Lys | Gln | |||||||||
| ON402789 VN/2021_Vietnam | Phe | Trp | Lys | Asn | Ile | Lys | Gln | |||||||||
| MW791760 2019-003-B_2019/Philippines | Phe | Trp | Lys | Asn | Ile | Lys | Gln | |||||||||
| ON963982 PHL/StrainA4_Philippines(2021) | Phe | X | X | X | X | X | X | |||||||||
| MW049116 pig/Yeoncheon1/2019_Korea(2019) | Phe | Trp | Lys | Asn | Ile | Lys | Gln | |||||||||
| OR159218 S-S-VR-413000-00008_Korea(2019) | Phe | Trp | Lys | Asn | Ile | Lys | Gln | |||||||||
| S. No. | Gene Name | Indels | Isolate Name |
|---|---|---|---|
| 1 | 110-13La-IGR-110-13Lb | 29 bp deletion | TH1_22/CR |
| 2 | ASFV G ACD 00350 | 52 bp deletion | TH1_22/CR |
| 3 | EP402R | 18 bp deletion | Vietnam (ON402789) |
| 4 | B475L | 78 bp addition | Vietnam (ON402789) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salman, M.; Venkateswaran, D.; Prakash, A.; Nguyen, Q.A.; Suntisukwattana, R.; Atthaapa, W.; Tantituvanont, A.; Songkasupa, T.; Deemagarn, T.; Bhakha, K.; et al. The Comparative Full-Length Genome Characterization of African Swine Fever Virus Detected in Thailand. Animals 2024, 14, 2602. https://doi.org/10.3390/ani14172602
Salman M, Venkateswaran D, Prakash A, Nguyen QA, Suntisukwattana R, Atthaapa W, Tantituvanont A, Songkasupa T, Deemagarn T, Bhakha K, et al. The Comparative Full-Length Genome Characterization of African Swine Fever Virus Detected in Thailand. Animals. 2024; 14(17):2602. https://doi.org/10.3390/ani14172602
Chicago/Turabian StyleSalman, Muhammad, Dhithya Venkateswaran, Anwesha Prakash, Quynh Anh Nguyen, Roypim Suntisukwattana, Waranya Atthaapa, Angkana Tantituvanont, Tapanut Songkasupa, Taweewat Deemagarn, Kultyarat Bhakha, and et al. 2024. "The Comparative Full-Length Genome Characterization of African Swine Fever Virus Detected in Thailand" Animals 14, no. 17: 2602. https://doi.org/10.3390/ani14172602
APA StyleSalman, M., Venkateswaran, D., Prakash, A., Nguyen, Q. A., Suntisukwattana, R., Atthaapa, W., Tantituvanont, A., Songkasupa, T., Deemagarn, T., Bhakha, K., Pengpetch, N., Saenboonrueng, J., Thaweerattanasinp, T., Jongkaewwattana, A., & Nilubol, D. (2024). The Comparative Full-Length Genome Characterization of African Swine Fever Virus Detected in Thailand. Animals, 14(17), 2602. https://doi.org/10.3390/ani14172602