
Sturnidae sensu lato Mitogenomics: Novel Insights into Codon Aversion, Selection, and Phylogeny
 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction, Sequencing, and Assembly
2.2. Gene Annotation and Comprehensive Sequence Analyses
2.3. Phylogenetic Inferences
3. Results
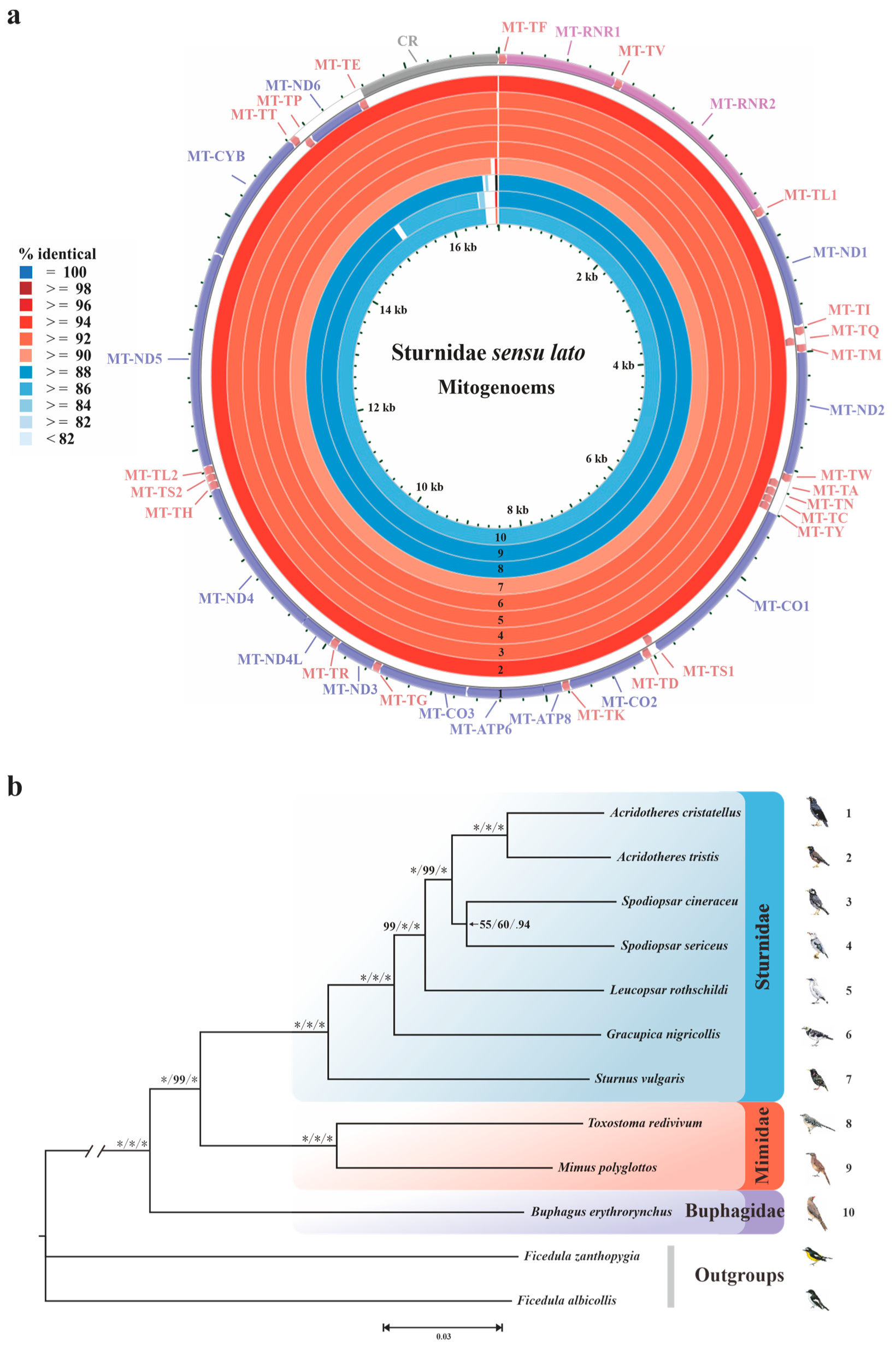
3.1. NGS Data Information and Mitogenome Organization
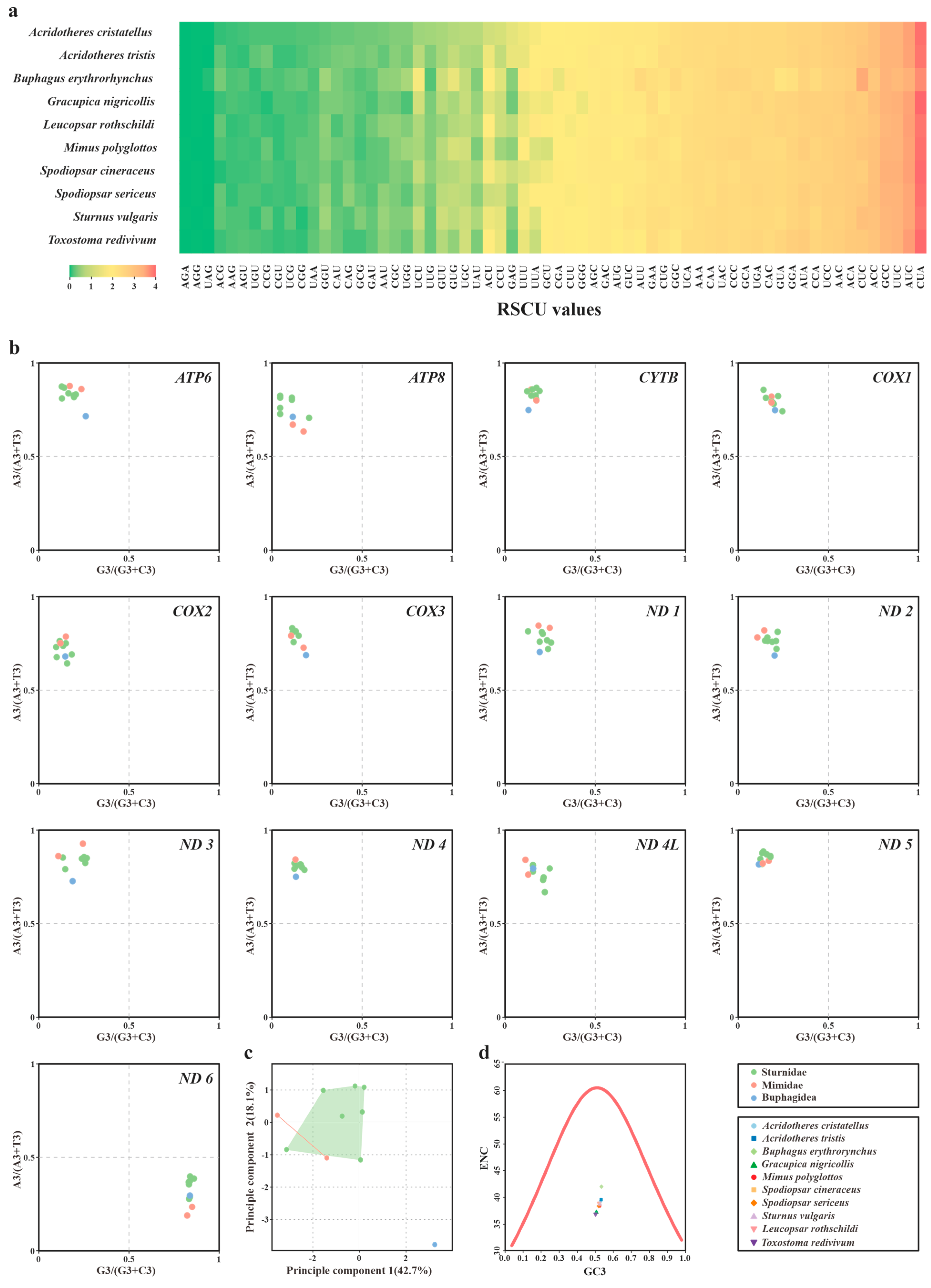
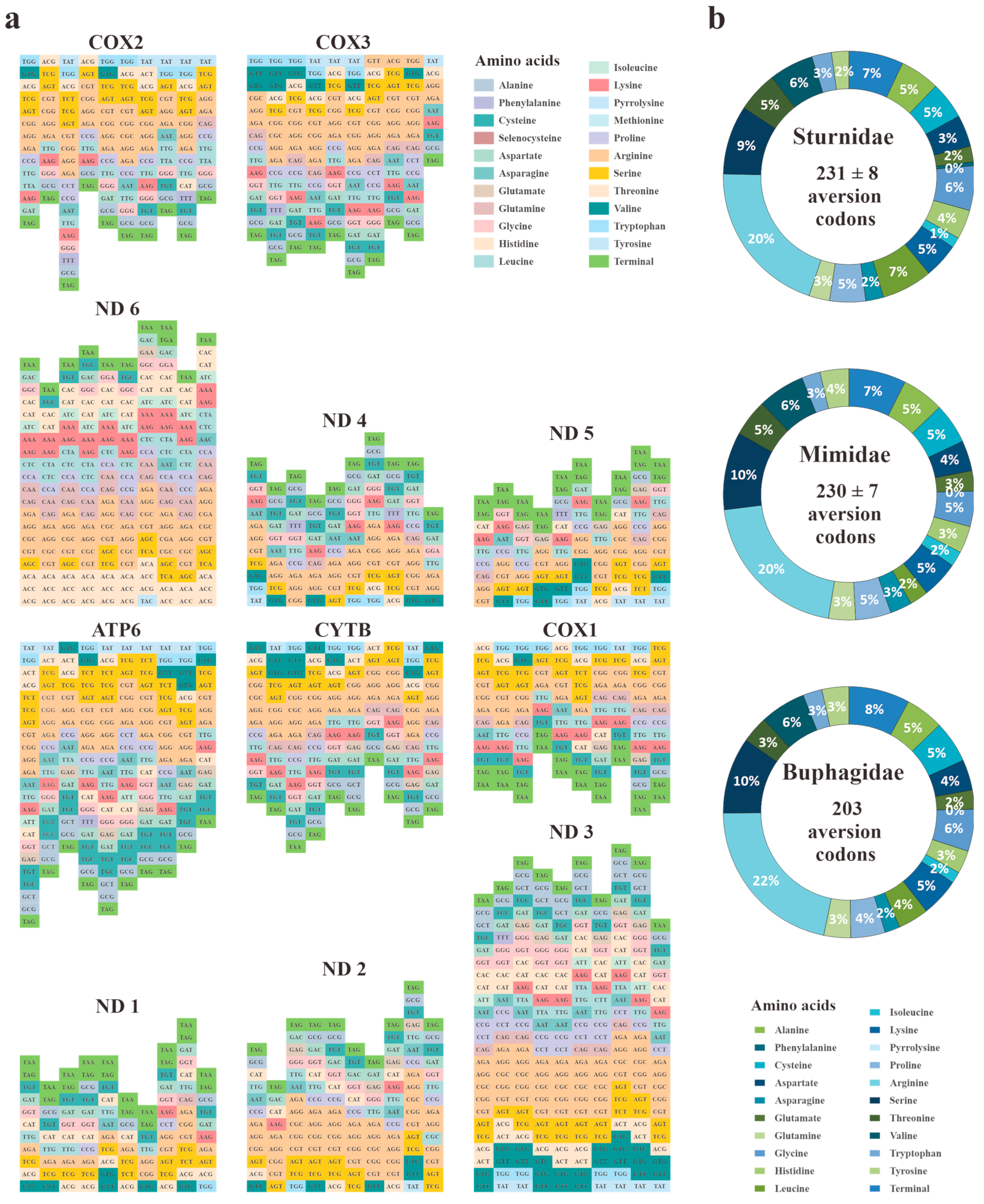
3.2. Codon Usage Bias (CUB) and Codon Aversion of Mitochondria PCGs
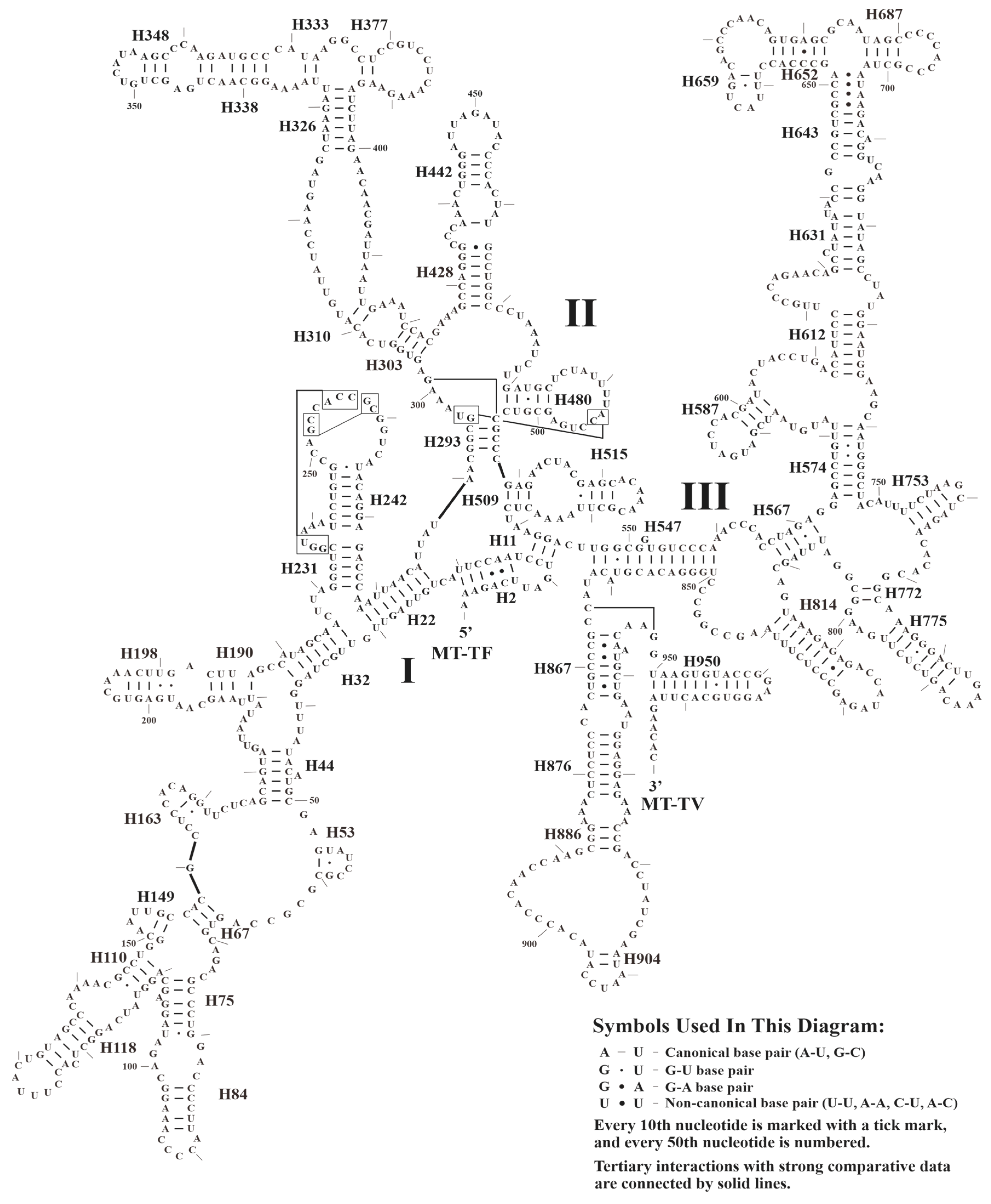
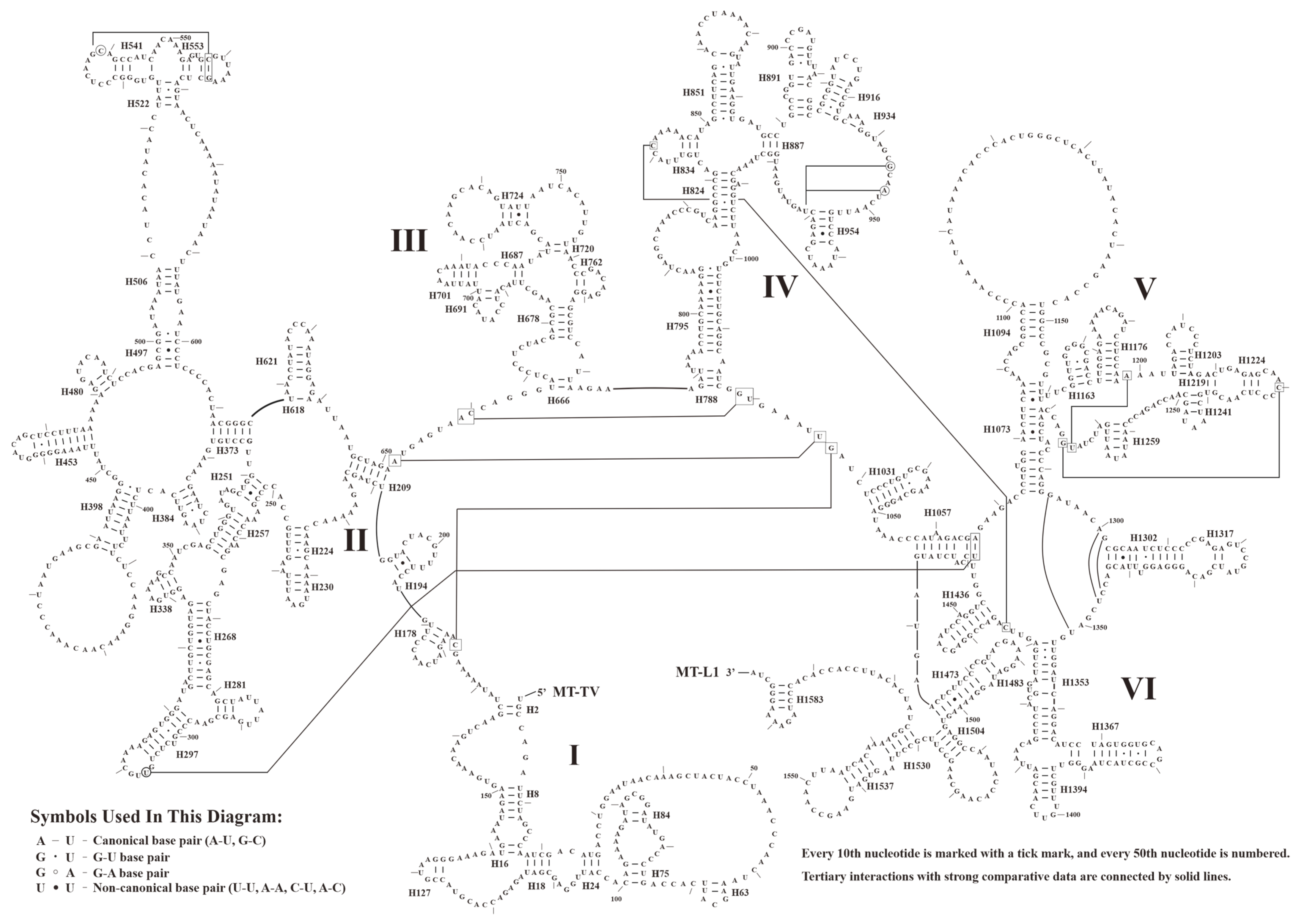
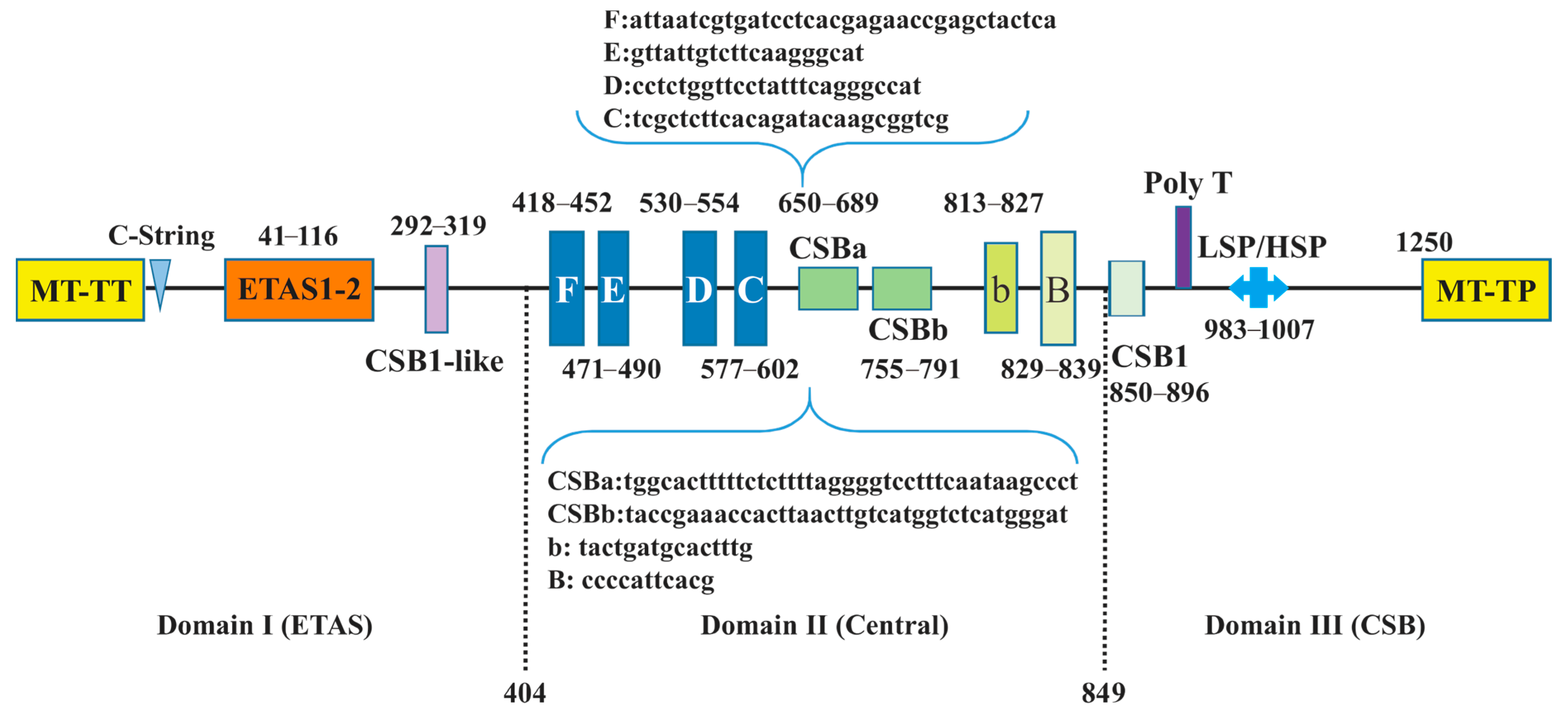
3.3. Secondary Structure of tRNAs, rRNAs, and CRs
3.4. Rates and Patterns of Mitochondrial Gene Evolution
3.5. Phylogenetic Implications
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cibois, A.; Cracraft, J. Assessing the passerine “Tapestry”: Phylogenetic relationships of the Muscicapoidea inferred from nuclear DNA sequences. Mol. Phylogenet. Evol. 2004, 32, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Lovette, I.J.; Rubenstein, D.R. A comprehensive molecular phylogeny of the starlings (Aves: Sturnidae) and mockingbirds (Aves: Mimidae): Congruent mtDNA and nuclear trees for a cosmopolitan avian radiation. Mol. Phylogenet. Evol. 2007, 44, 1031–1056. [Google Scholar] [CrossRef] [PubMed]
- Zuccon, D.; Cibois, A.; Pasquet, E.; Ericson, P.G. Nuclear and mitochondrial sequence data reveal the major lineages of starlings, mynas and related taxa. Mol. Phylogenet. Evol. 2006, 41, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.F.; Schulenberg, T.S.; Iliff, M.J.; Billerman, S.M.; Fredericks, T.A.; Sullivan, B.L.; Wood, C.L. The eBird/Clements Checklist of Birds of the World: V2019. 2019. Available online: https://www.birds.cornell.edu/clementschecklist/download/ (accessed on 25 June 2021).
- Mengesha, Y.A. A study of oxpecker-mammal symbiosis in Ethiopia. East Afr. Agric. For. J. 1978, 43, 321–326. [Google Scholar] [CrossRef]
- Jubber, W.R. Interspecific competition between Oxpecker Buphagus species. Biodivers. Observ. 2014, 5, 325–335. [Google Scholar]
- DaCosta, J.M.; Miller, M.J.; Mortensen, J.L.; Reed, J.M.; Curry, R.L.; Sorenson, M.D. Phylogenomics clarifies biogeographic and evolutionary history, and conservation status of West Indian tremblers and thrashers (Aves: Mimidae). Mol. Phylogenet. Evol. 2019, 136, 196–205. [Google Scholar] [CrossRef]
- Lovette, I.J.; Arbogast, B.S.; Curry, R.L.; Zink, R.M.; Botero, C.A.; Sullivan, J.P.; Talaba, A.L.; Harris, R.B.; Rubenstein, D.R.; Ricklefs, R.E. Phylogenetic relationships of the mockingbirds and thrashers (Aves: Mimidae). Mol. Phylogenet. Evol. 2012, 63, 219–229. [Google Scholar] [CrossRef]
- Lovette, I.J.; McCleery, B.V.; Talaba, A.L.; Rubenstein, D.R. A complete species-level molecular phylogeny for the “Eurasian” starlings (Sturnidae: Sturnus, Acridotheres, and allies): Recent diversification in a highly social and dispersive avian group. Mol. Phylogenet. Evol. 2008, 47, 251–260. [Google Scholar] [CrossRef]
- Zuccon, D.; Pasquet, E.; Ericson, P.G. Phylogenetic relationships among Palearctic–Oriental starlings and mynas (genera Sturnus and Acridotheres: Sturnidae). Zool. Scr. 2008, 37, 469–481. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Green, D.R. Apoptotic pathways: The roads to ruin. Cell 1998, 94, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed]
- Tiku, V.; Tan, M.-W.; Dikic, I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Yahalomi, D.; Atkinson, S.D.; Neuhof, M.; Chang, E.S.; Philippe, H.; Cartwright, P.; Bartholomew, J.L.; Huchon, D. A cnidarian parasite of salmon (Myxozoa: Henneguya) lacks a mitochondrial genome. Proc. Natl. Acad. Sci. USA 2020, 117, 5358–5363. [Google Scholar] [CrossRef]
- Desjardins, P.; Morais, R. Sequence and gene organization of the chicken mitochondrial genome: A novel gene order in higher vertebrates. J. Mol. Biol. 1990, 212, 599–634. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Yuan, J.; Ren, L.; Zhang, L.; Zhang, L.; Jiang, L.; Chen, D.; Kan, X.; Zhang, B. The complete mitochondrial genome of the yellow-browed bunting, Emberiza chrysophrys (Passeriformes: Emberizidae), and phylogenetic relationships within the genus Emberiza. J. Genet. 2014, 93, 699–707. [Google Scholar] [CrossRef]
- Dai, L.; Zhou, X.; Kausar, S.; Abbas, M.N.; Wu, L.; Zhou, H. Mitochondrial genome of Diaphania indica (saunders) (Lepidoptera: Pyraloidea) and implications for its phylogeny. Int. J. Biol. Macromol. 2018, 108, 981–989. [Google Scholar] [CrossRef]
- Sriboonlert, A.; Wonnapinij, P. Comparative mitochondrial genome analysis of the firefly, Inflata indica (Coleoptera: Lampyridae) and the first evidence of heteroplasmy in fireflies. Int. J. Biol. Macromol. 2019, 121, 671–676. [Google Scholar] [CrossRef]
- Yuan, X.; Cao, M.; Li, P.; Cheng, S.; Liu, X.; Du, Y.; Zhang, Z.; Shen, G.; Zhang, P. The mitochondrial genome of Arthrinium arundinis and its phylogenetic position within Sordariomycetes. Int. J. Biol. Macromol. 2019, 121, 956–963. [Google Scholar] [CrossRef]
- Su, T.; Liang, A. Comparative analysis of seven mitochondrial genomes of Phymatostetha (Hemiptera: Cercopidae) and phylogenetic implications. Int. J. Biol. Macromol. 2019, 125, 1112–1117. [Google Scholar] [CrossRef]
- Curole, J.P.; Kocher, T.D. Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 1999, 14, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Bi, D.; Han, S.; Yi, R.; Zhang, S.; Ye, Y.; Gao, J.; Yang, J.; Kan, X. Mitogenomic Codon Usage Patterns of Superfamily Certhioidea (Aves, Passeriformes): Insights into Asymmetrical Bias and Phylogenetic Implications. Animals 2022, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Barker, F.K. Mitogenomic data resolve basal relationships among passeriform and passeridan birds. Mol. Phylogenet. Evol. 2014, 79, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Bi, D.; Ding, H.; Wang, Q.; Jiang, L.; Lu, W.; Wu, X.; Zhu, R.; Zeng, J.; Zhou, S.; Yang, X. Two new mitogenomes of Picidae (Aves, Piciformes): Sequence, structure and phylogenetic analyses. Int. J. Biol. Macromol. 2019, 133, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Gibb, G.C.; Kardailsky, O.; Kimball, R.T.; Braun, E.L.; Penny, D. Mitochondrial genomes and avian phylogeny: Complex characters and resolvability without explosive radiations. Mol. Biol. Evol. 2007, 24, 269–280. [Google Scholar] [CrossRef]
- Huang, Z.; Tu, F.; Tang, S. Two new mitogenomes of Pellorneidae (Aves: Passeriformes) and a phylogeny of the superfamily Sylvioidea. Aust. J. Zool. 2019, 66, 167–173. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, J.; Wang, P.; Ren, Q.; Yuan, J.; Qian, C.; Hua, X.; Guo, Z.; Zhang, L.; Yang, J. The mitochondrial genomes of Aquila fasciata and Buteo lagopus (Aves, Accipitriformes): Sequence, structure and phylogenetic analyses. PLoS ONE 2015, 10, e0136297. [Google Scholar] [CrossRef]
- Kan, X.Z.; Li, X.F.; Zhang, L.Q.; Chen, L.; Qian, C.J.; Zhang, X.W.; Wang, L. Characterization of the complete mitochondrial genome of the Rock pigeon, Columba livia (Columbiformes: Columbidae). Genet. Mol. Res. 2010, 9, 1234–1249. [Google Scholar] [CrossRef]
- Kan, X.Z.; Yang, J.K.; Li, X.F.; Chen, L.; Lei, Z.P.; Wang, M.; Qian, C.J.; Gao, H.; Yang, Z.Y. Phylogeny of major lineages of galliform birds (Aves: Galliformes) based on complete mitochondrial genomes. Genet. Mol. Res. 2010, 9, 1625–1633. [Google Scholar] [CrossRef]
- Mackiewicz, P.; Urantówka, A.D.; Kroczak, A.; Mackiewicz, D. Resolving phylogenetic relationships within Passeriformes based on mitochondrial genes and inferring the evolution of their mitogenomes in terms of duplications. Genome Biol. Evol. 2019, 11, 2824–2849. [Google Scholar] [CrossRef]
- Marshall, H.D.; Baker, A.J.; Grant, A.R. Complete mitochondrial genomes from four subspecies of common chaffinch (Fringilla coelebs): New inferences about mitochondrial rate heterogeneity, neutral theory, and phylogenetic relationships within the order Passeriformes. Gene 2013, 517, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Huang, J.; Yan, C.; Xu, G.; Zhang, X.; Yue, B. The complete mitochondrial genome of Accipiter virgatus and evolutionary history of the pseudo-control regions in Falconiformes. Biochem. Syst. Ecol. 2015, 58, 75–84. [Google Scholar] [CrossRef]
- Sun, C.-H.; Liu, H.-Y.; Lu, C.-H. Five new mitogenomes of Phylloscopus (Passeriformes, Phylloscopidae): Sequence, structure, and phylogenetic analyses. Int. J. Biol. Macromol. 2020, 146, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Pan, T.; Hu, C.; Sun, L.; Ding, H.; Wang, H.; Zhang, C.; Jin, H.; Chang, Q.; Kan, X. Rapid and recent diversification patterns in Anseriformes birds: Inferred from molecular phylogeny and diversification analyses. PLoS ONE 2017, 12, e0184529. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, L.; Gowda, V.; Wang, M.; Li, X.; Kan, X. The mitochondrial genome of the Cinnamon Bittern, Ixobrychus cinnamomeus (Pelecaniformes: Ardeidae): Sequence, structure and phylogenetic analysis. Mol. Biol. Rep. 2012, 39, 8315–8326. [Google Scholar] [CrossRef]
- Zhou, X.; Lin, Q.; Fang, W.; Chen, X. The complete mitochondrial genomes of sixteen ardeid birds revealing the evolutionary process of the gene rearrangements. BMC Genom. 2014, 15, 573. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; de Pamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Braschi, B.; Denny, P.; Gray, K.; Jones, T.; Seal, R.; Tweedie, S.; Yates, B.; Bruford, E. Genenames. org: The HGNC and VGNC resources in 2019. Nucleic Acids Res. 2018, 47, D786–D792. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Arantes, A.S.; Stothard, P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics 2012, 13, 202. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Peden, J.F. Analysis of Codon Usage; Citeseer: Princeton, NJ, USA, 2000. [Google Scholar]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Li, X.; Huang, Y.; Lei, F. Comparative mitochondrial genomics and phylogenetic relationships of the Crossoptilon species (Phasianidae, Galliformes). BMC Genom. 2015, 16, 42. [Google Scholar] [CrossRef]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D’Souza, L.M.; Du, Y.; Feng, B.; Lin, N.; Madabusi, L.V.; Müller, K.M. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform. 2002, 3, 2. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Swofford, D.L. Phylogenetic Analysis Using Parsimony (* and other Methods); Sinauer Associate: Sunderland, MA, USA, 2002. [Google Scholar]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Sangster, G.; Luksenburg, J.A. Sharp increase of problematic mitogenomes of birds: Causes, consequences, and remedies. Genome Biol. Evol. 2021, 13, evab210. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Stiller, J.; Deng, Y.; Armstrong, J.; Fang, Q.; Reeve, A.H.; Xie, D.; Chen, G.; Guo, C.; Faircloth, B.C. Dense sampling of bird diversity increases power of comparative genomics. Nature 2020, 587, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Ren, Q.; Kan, X.; Guo, Z.; Yang, J.; Li, X.; Yuan, J.; Qian, M.; Hu, Q.; Zhang, L. Complete mitochondrial genome of the Red-billed Starling, Sturnus sericeus (Aves: Passeriformes): The first representative of the family Sturnidae with a single control region. Mitochondrial DNA 2013, 24, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Rollins, L.A.; Woolnough, A.P.; Fanson, B.G.; Cummins, M.L.; Crowley, T.M.; Wilton, A.N.; Sinclair, R.; Butler, A.; Sherwin, W.B. Selection on mitochondrial variants occurs between and within individuals in an expanding invasion. Mol. Biol. Evol. 2016, 33, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.J.F.; Feare, C.J. Sharpe’s Starling (Pholia sharpii), version 1.0. In Birds of the World; del Hoyo, J., Elliott, A., Sargatal, J., Christie, D.A., de Juana, E., Eds.; Cornell Lab of Ornithology: Ithaca, NY, USA, 2020. [Google Scholar] [CrossRef]
- Gill, F.; Donsker, D.; Rasmussen, P. IOC World Bird List (v11.1). 2021. Available online: https://www.worldbirdnames.org/new/ (accessed on 26 June 2021).
- Yang, J.; Ding, H.; Kan, X. Codon usage patterns and evolution of HSP60 in birds. Int. J. Biol. Macromol. 2021, 183, 1002–1012. [Google Scholar] [CrossRef]
- Iriarte, A.; Lamolle, G.; Musto, H. Codon Usage Bias: An Endless Tale. J. Mol. Evol. 2021, 89, 589–593. [Google Scholar] [CrossRef]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Ismail, S.N.F.B.; Baharum, S.N.; Fazry, S.; Low, C.F. Comparative genome analysis reveals a distinct influence of nucleotide composition on virus–host species-specific interaction of prawn-infecting nodavirus. J. Fish Dis. 2019, 42, 1761–1772. [Google Scholar] [CrossRef]
- Yuan, M.L.; Zhang, L.J.; Zhang, Q.L.; Zhang, L.; Li, M.; Wang, X.T.; Feng, R.Q.; Tang, P.A. Mitogenome evolution in ladybirds: Potential association with dietary adaptation. Ecol. Evol. 2020, 10, 1042–1053. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, J.; Qiao, J.; Li, W.; Li, J.; Xu, R.; Wang, H.; Liu, Z.; Xing, B.; Wendel, J.F. Comparative analysis of codon usage between Gossypium hirsutum and G. barbadense mitochondrial genomes. Mitochondrial DNA B 2020, 5, 2500–2506. [Google Scholar] [CrossRef]
- Guan, D.-L.; Qian, Z.-Q.; Ma, L.-B.; Bai, Y.; Xu, S.-Q. Different mitogenomic codon usage patterns between damselflies and dragonflies and nine complete mitogenomes for odonates. Sci. Rep. 2019, 9, 678. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; He, J.; Jia, X.; Qi, Q.; Liang, Z.; Zheng, H.; Ping, Y.; Liu, S.; Sun, J. Analysis of codon usage bias of mitochondrial genome in Bombyx moriand its relation to evolution. BMC Evol. Biol. 2014, 14, 262. [Google Scholar] [CrossRef] [PubMed]
- Forcelloni, S.; Giansanti, A. Evolutionary Forces and Codon Bias in Different Flavors of Intrinsic Disorder in the Human Proteome. J. Mol. Evol. 2020, 88, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.B.; Hippen, A.A.; Belyeu, J.R.; Whiting, M.F.; Ridge, P.G. Missing something? Codon aversion as a new character system in phylogenetics. Cladistics 2017, 33, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.B.; McKinnon, L.M.; Whiting, M.F.; Ridge, P.G. Codon use and aversion is largely phylogenetically conserved across the tree of life. Mol. Phylogenet. Evol. 2020, 144, 106697. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Bi, D.; Yi, R.; Ding, H.; Wu, L.; Kan, X. Plastome evolution of Aeonium and Monanthes (Crassulaceae): Insights into the variation of plastomic tRNAs, and the patterns of codon usage and aversion. Planta 2022, 256, 35. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Han, S.; Ye, Y.; Bi, D.; Zhang, S.; Yi, R.; Gao, J.; Yang, J.; Wu, L.; Kan, X. Ten Plastomes of Crassula (Crassulaceae) and Phylogenetic Implications. Biology 2022, 11, 1779. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Wang, R.; Hong, X.; Wu, C.; Zhang, S.; Kan, X. Plastomes of Bletilla (Orchidaceae) and Phylogenetic Implications. Int. J. Mol. Sci. 2022, 23, 10151. [Google Scholar] [CrossRef]
- Yarham, J.W.; Elson, J.L.; Blakely, E.L.; McFarland, R.; Taylor, R.W. Mitochondrial tRNA mutations and disease. Wiley Interdiscip. Rev. RNA 2010, 1, 304–324. [Google Scholar] [CrossRef]
- Kim, S.; Sussman, J.; Suddath, F.; Quigley, G.; McPherson, A.; Wang, A.; Seeman, N.; Rich, A. The general structure of transfer RNA molecules. Proc. Natl. Acad. Sci. USA 1974, 71, 4970–4974. [Google Scholar] [CrossRef]
- Hanada, T.; Suzuki, T.; Watanabe, K. Translation activity of mitochondrial tRNA with unusual secondary structure. In Nucleic Acids Symposium Series; Oxford University Press: Oxford, UK, 2000; pp. 249–250. [Google Scholar]
- Hanada, T.; Suzuki, T.; Yokogawa, T.; Takemoto-Hori, C.; Sprinzl, M.; Watanabe, K. Translation ability of mitochondrial tRNAsSer with unusual secondary structures in an in vitro translation system of bovine mitochondria. Genes Cells 2001, 6, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, T.; Sato, A.; Sakurai, M.; Watanabe, K.; Ohtsuki, T. A unique tRNA recognition mechanism of Caenorhabditis elegans mitochondrial EF-Tu2. Nucleic Acids Res. 2005, 33, 4683–4691. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Kawai, G.; Watanabe, K. The minimal tRNA: Unique structure of Ascaris suum mitochondrial tRNASerUCU having a short T arm and lacking the entire D arm. FEBS Lett. 2002, 514, 37–43. [Google Scholar] [CrossRef]
- Frazer-Abel, A.A.; Hagerman, P.J. Variation of the acceptor–anticodon interstem angles among mitochondrial and non-mitochondrial tRNAs. J. Mol. Biol. 2004, 343, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012, 40, 2833–2845. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.-i.; Suematsu, T.; Ohtsuki, T. Losing the stem-loop structure from metazoan mitochondrial tRNAs and co-evolution of interacting factors. Front. Genet. 2014, 5, 109. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wang, Q.-X.; Huang, Y.; Xiao, H. Analysis of the complete mitochondrial genome sequence of Larus brunnicephalus (Aves, Laridae). Yi Chuan = Hereditas 2012, 34, 1434–1446. [Google Scholar] [CrossRef]
- Harrison, G.L.; McLenachan, P.A.; Phillips, M.J.; Slack, K.E.; Cooper, A.; Penny, D. Four new avian mitochondrial genomes help get to basic evolutionary questions in the late cretaceous. Mol. Biol. Evol. 2004, 21, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.G.; Huang, Y.; Lei, F.M. Sequencing and phylogenetic analysis of the Pyrgilauda ruficollis (Aves, Passeridae) complete mitochondrial genome. Dongwuxue Yanjiu 2014, 35, 81–91. [Google Scholar]
- Torres, A.; Goloboff, P.A.; Catalano, S.A. Assessing topological congruence among concatenation-based phylogenomic approaches in empirical datasets. Mol. Phylogenet. Evol. 2021, 161, 107086. [Google Scholar] [CrossRef]
- Hunt, J.S.; Bermingham, E.; Ricklefs, E.R. Molecular systematics and biogeography of Antillean thrashers, tremblers, and mockingbirds (Aves: Mimidae). Auk 2001, 118, 35–55. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Accession | Reference | Size (bp)/AT% | |||||
|---|---|---|---|---|---|---|---|---|---|
| Heavy-Strand | PCGs | MT-RNR1 | MT-RNR2 | tRNA Genes | CR | ||||
| Buphagidae | Buphagus erythrorynchus | MN356312 | [56] | 16,802/51.85 | 11,400/50.65 | 979/50.97 | 1602/54.24 | 1546/57.18 | 1231/54.51 |
| Mimidae | Mimus polyglottos | BK010995 # | This study | 16,804/52.46 | 11,400/51.26 | 985/51.37 | 1600/54.44 | 1540/57.27 | 1231/56.38 |
| Mimidae | Toxostoma redivivum | MN356247 | [56] | 16,780/52.91 | 11,400/52.13 | 983/50.76 | 1600/54.81 | 1542/56.49 | 1208/55.55 |
| Sturnidae | Acridotheres cristatellus | NC_015613 | This study | 16,820/52.34 | 11,400/51.18 | 979/50.97 | 1598/54.76 | 1541/57.30 | 1250/55.52 |
| Sturnidae | Acridotheres tristis | NC_015195 | This study | 16,822/51.99 | 11,400/50.87 | 979/50.97 | 1598/54.38 | 1542/57.00 | 1251/54.60 |
| Sturnidae | Gracupica nigricollis | NC_020423 | This study | 16,839/52.95 | 11,400/51.96 | 978/50.92 | 1602/55.06 | 1541/58.08 | 1253/55.55 |
| Sturnidae | Leucopsar rothschildi | MN356237 | [56] | 16,845/52.70 | 11,400/51.61 | 978/51.53 | 1600/55.13 | 1543/57.23 | 1254/55.58 |
| Sturnidae | Spodiopsar cineraceus | NC_015237 | This study | 16,821/52.49 | 11,400/51.29 | 980/51.53 | 1598/55.19 | 1541/57.82 | 1249/54.68 |
| Sturnidae | Spodiopsar sericeus | NC_014455 | [57] | 16,823/52.67 | 11,400/51.51 | 979/51.69 | 1598/55.07 | 1543/57.23 | 1250/55.76 |
| Sturnidae | Sturnus vulgaris | NC_029360 | [58] | 16,793/52.38 | 11,400/51.29 | 974/50.72 | 1599/54.28 | 1540/57.60 | 1233/55.31 |
| Gene | Length (bp) | Var. Sites [%] | π | dN | dS | dN/dS | ts/tv |
|---|---|---|---|---|---|---|---|
| ATP6 | 684 | 224 (32.75) | 0.11816 | 0.1127 | 5.4502 | 0.02068 | 10.2768 |
| ATP8 | 168 | 58 (34.52) | 0.13294 | 0.433 | 1.6895 | 0.25631 | 5.4548 |
| CO1 | 1551 | 369 (23.79) | 0.09015 | 0.0191 | 3.3076 | 0.00579 | 6.1603 |
| CO2 | 684 | 196 (28.65) | 0.10679 | 0.0855 | 3.9109 | 0.02185 | 6.6734 |
| CO3 | 786 | 215 (27.35) | 0.0955 | 0.0756 | 4.1272 | 0.01833 | 7.3630 |
| CYB | 1143 | 292 (25.55) | 0.10192 | 0.0844 | 4.7612 | 0.01772 | 6.4004 |
| ND1 | 978 | 330 (33.74) | 0.13263 | 0.0848 | 4.9668 | 0.01708 | 9.2547 |
| ND2 | 1041 | 392 (37.66) | 0.13797 | 0.2108 | 4.1455 | 0.05085 | 8.5414 |
| ND3 | 351 | 115 (32.76) | 0.12023 | 0.1351 | 3.7409 | 0.03611 | 8.9541 |
| ND4 | 1380 | 457 (33.12) | 0.12308 | 0.1077 | 7.2908 | 0.01477 | 12.7131 |
| ND4L | 297 | 97 (32.66) | 0.1138 | 0.0906 | 7.2069 | 0.01257 | 11.8799 |
| ND5 | 1818 | 570 (31.35) | 0.10785 | 0.1326 | 5.9385 | 0.02232 | 9.6700 |
| MT-ND6 | 519 | 178 (34.30) | 0.13697 | 0.2388 | 2.5386 | 0.09406 | 6.3762 |
| Overall | 11,400 | 3493 (30.64) | 0.1135 |
| Gene | Model | Model Setting | |
|---|---|---|---|
| Lset nst | Rate | ||
| MT-ATP6 | HKY+I+G4 | 2 | invgamma |
| MT-ATP8 | GTR+I | 6 | propinv |
| MT-CO1 | GTR+G4 | 6 | gamma |
| MT-CO2 | HKY+I+G4 | 2 | invgamma |
| MT-CO3 | HKY+G4 | 2 | gamma |
| MT-CYB | HKY+I+G4 | 2 | invgamma |
| MT-ND1 | HKY+I+G4 | 2 | invgamma |
| MT-ND2 | HKY+I+G4 | 2 | invgamma |
| MT-ND3 | HKY+G4 | 2 | gamma |
| MT-ND4 | GTR+G4 | 6 | gamma |
| MT-ND4L | HKY+G4 | 2 | gamma |
| MT-ND5 | HKY+I+G4 | 2 | invgamma |
| MT-ND6 | HKY+I+G4 | 2 | invgamma |
| Gene | Codon Position | Iss a | Iss.cSym b | p | Iss.cAsym c | p |
|---|---|---|---|---|---|---|
| ATP6 | 1st | 0.0877 | 0.6828 | <0.0001 | 0.5256 | <0.0001 |
| 2nd | 0.0201 | 0.6828 | <0.0001 | 0.5256 | <0.0001 | |
| 3rd | 0.486 | 0.6828 | <0.0001 | 0.5256 | 0.1973 | |
| ATP8 | 1st | 0.1931 | 0.8013 | <0.0001 | 0.8228 | <0.0001 |
| 2nd | 0.1114 | 0.8013 | <0.0001 | 0.8228 | <0.0001 | |
| 3rd | 0.3077 | 0.8013 | <0.0001 | 0.8228 | <0.0001 | |
| CO1 | 1st | 0.0202 | 0.7237 | <0.0001 | 0.5571 | <0.0001 |
| 2nd | 0.0026 | 0.7237 | <0.0001 | 0.5571 | <0.0001 | |
| 3rd | 0.4206 | 0.7237 | <0.0001 | 0.5571 | <0.0001 | |
| CO2 | 1st | 0.0478 | 0.6828 | <0.0001 | 0.5256 | <0.0001 |
| 2nd | 0.0139 | 0.6828 | <0.0001 | 0.5256 | <0.0001 | |
| 3rd | 0.4466 | 0.6828 | <0.0001 | 0.5256 | 0.0125 | |
| CO3 | 1st | 0.0407 | 0.6873 | <0.0001 | 0.526 | <0.0001 |
| 2nd | 0.0236 | 0.6873 | <0.0001 | 0.526 | <0.0001 | |
| 3rd | 0.444 | 0.6873 | <0.0001 | 0.526 | 0.0049 | |
| CYB | 1st | 0.0609 | 0.7051 | <0.0001 | 0.5386 | <0.0001 |
| 2nd | 0.0088 | 0.7051 | <0.0001 | 0.5386 | <0.0001 | |
| 3rd | 0.4611 | 0.7051 | <0.0001 | 0.5386 | 0.0042 | |
| ND1 | 1st | 0.0734 | 0.6969 | <0.0001 | 0.5316 | <0.0001 |
| 2nd | 0.0168 | 0.6969 | <0.0001 | 0.5316 | <0.0001 | |
| 3rd | 0.4984 | 0.6969 | <0.0001 | 0.5316 | 0.1917 | |
| ND2 | 1st | 0.1217 | 0.7001 | <0.0001 | 0.5341 | <0.0001 |
| 2nd | 0.0524 | 0.7001 | <0.0001 | 0.5341 | <0.0001 | |
| 3rd | 0.4747 | 0.7001 | <0.0001 | 0.5341 | 0.0189 | |
| ND3 | 1st | 0.1075 | 0.6886 | <0.0001 | 0.5759 | <0.0001 |
| 2nd | 0.0635 | 0.6886 | <0.0001 | 0.5759 | <0.0001 | |
| 3rd | 0.4817 | 0.6886 | 0.0001 | 0.5759 | 0.0643 | |
| ND4 | 1st | 0.0871 | 0.7161 | <0.0001 | 0.5494 | <0.0001 |
| 2nd | 0.03 | 0.7161 | <0.0001 | 0.5494 | <0.0001 | |
| 3rd | 0.4936 | 0.7161 | <0.0001 | 0.5494 | 0.011 | |
| ND4L | 1st | 0.0978 | 0.7016 | <0.0001 | 0.6103 | <0.0001 |
| 2nd | 0.0203 | 0.7016 | <0.0001 | 0.6103 | <0.0001 | |
| 3rd | 0.4469 | 0.7016 | <0.0001 | 0.6103 | 0.0005 | |
| ND5 | 1st | 0.0802 | 0.7349 | <0.0001 | 0.5684 | <0.0001 |
| 2nd | 0.0374 | 0.7349 | <0.0001 | 0.5684 | <0.0001 | |
| 3rd | 0.4608 | 0.7349 | <0.0001 | 0.5684 | <0.0001 | |
| ND6 | 1st | 0.1664 | 0.6787 | <0.0001 | 0.5345 | <0.0001 |
| 2nd | 0.0689 | 0.6787 | <0.0001 | 0.5345 | <0.0001 | |
| 3rd | 0.4371 | 0.6787 | <0.0001 | 0.5345 | 0.0135 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.; Ding, H.; Peng, H.; Dai, C.; Zhang, S.; Yang, J.; Gao, J.; Kan, X. Sturnidae sensu lato Mitogenomics: Novel Insights into Codon Aversion, Selection, and Phylogeny. Animals 2024, 14, 2777. https://doi.org/10.3390/ani14192777
Han S, Ding H, Peng H, Dai C, Zhang S, Yang J, Gao J, Kan X. Sturnidae sensu lato Mitogenomics: Novel Insights into Codon Aversion, Selection, and Phylogeny. Animals. 2024; 14(19):2777. https://doi.org/10.3390/ani14192777
Chicago/Turabian StyleHan, Shiyun, Hengwu Ding, Hui Peng, Chenwei Dai, Sijia Zhang, Jianke Yang, Jinming Gao, and Xianzhao Kan. 2024. "Sturnidae sensu lato Mitogenomics: Novel Insights into Codon Aversion, Selection, and Phylogeny" Animals 14, no. 19: 2777. https://doi.org/10.3390/ani14192777
APA StyleHan, S., Ding, H., Peng, H., Dai, C., Zhang, S., Yang, J., Gao, J., & Kan, X. (2024). Sturnidae sensu lato Mitogenomics: Novel Insights into Codon Aversion, Selection, and Phylogeny. Animals, 14(19), 2777. https://doi.org/10.3390/ani14192777