Simple Summary
The present study aimed to assemble and annotate the mitochondrial genome of Keenocardium buelowi and conducted an in-depth analysis to investigate the molecular features of this mitochondrial genome. Our findings established phylogenetic relationships within the Cardiidae family. Additionally, evidence of selection pressure on the cytochrome b gene was detected, suggesting its important role in the evolution of K. buelowi.
Abstract
The mitochondrial genome provides valuable data for phylogenetic analysis and evolutionary research. In this study, we sequenced, assembled, and annotated the mitochondrial genome of Keenocardium buelowi using the Illumina platform. The genome spanned 16,967 bp and included 13 protein-coding genes (PCGs), two ribosomal RNAs, and 22 transfer RNAs. All PCGs utilized standard ATN start codons and TAN stop codons. The phylogenetic tree based on maximum likelihood and Bayesian inference analyses revealed Clinocardiinae as the sister group to Trachycardiinae, with the estimated divergence time being 44.5 million years ago (MYA) between K. buelowi and Vasticardium flavum. Notably, the cytochrome b gene (cob) exhibited a positive selection signal. Our findings provide valuable insights into the evolutionary history and molecular phylogeny of K. buelowi.
1. Introduction
Keenocardium buelowi, a marine bivalve belonging to the Cardiidae family, is found at a depth of 10–50 m and is distributed across the eastern, southern, and western seas of the Korean Peninsula [1]. In Korea, this cockle species is predominantly collected through coastal fishing. Moreover, K. buelowi can be distinguished by its yellow foot, in contrast to the reddish foot found in Tegillarca granosa [2]. As these cockles are either consumed by local residents upon capture or unrecorded by fishermen, their reported yield is very low. Therefore, comprehensive research and data on K. buelowi are limited. In contrast, other cockle species, such as T. granosa [3] and Fulvia mutica [4], are extensively harvested in South Korea. In 2017, approximately 115 tons of T. granosa were harvested in South Korea [3]. The harvest season for cockles typically extends from late autumn to early spring [5]. However, the optimal period for harvesting K. buelowi extends from early summer to late autumn. Recently, the documented harvested biomass of harvested K. buelowi has been increasing, particularly in the southern sea of Korea, highlighting their potential as a valuable fishery resource and an alternative to expensive cockles.
Despite the increasing interest in K. buelowi, a significant gap exists in the genomic data necessary to fully understand its biological and ecological characteristics. To address this gap, we performed a detailed analysis of the mitochondrial genome of K. buelowi. In animals, mitochondria exhibit several unique characteristics. In particular, they are typically maternally inherited, highly conserved, and present in multiple copies in a cell. Moreover, they have a low rate of sequence recombination and evolve rapidly [6]. Because of these characteristics, mitochondrial sequences are particularly valuable for phylogenetic research [7]. Despite the extensive application of mitochondrial genome sequences in various studies, no complete mitochondrial sequences from the Clinocardiinae subfamily are publicly available as of July 2024. Additionally, only 13 complete mitochondrial genomes from seven genera were available.
In this study, we employed high-throughput sequencing technology (next-generation sequencing) to assemble the complete mitochondrial genome of K. buelowi. Furthermore, we compared the mitochondrial genome of K. buelowi with those of other Cardiidae species to infer their phylogenetic relationships and elucidate their evolutionary histories. Our findings may enhance our understanding of the evolutionary dynamics of K. buelowi, providing a foundation for future research and the sustainable management of this potentially valuable fishery resource.
2. Materials and Methods
2.1. Sample Collection and Sequencing
A healthy individual sample of K. buelowi was collected from Yokjido Island (35°3′29′′ N, 127°44′59′′ E), South Korea, in March 2024 using a shellfish dredge. The sample was then transported to the laboratory. Subsequently, the foot tissue was dissected, preserved in 99% ethanol, and stored in a −80 °C freezer.
Whole genomic DNA was extracted using the QIAGEN Blood and Cell Culture DNA Mini Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. A 150-bp paired-end library was then generated using the TruSeq DNA Nano 550 Kit (Illumina, Inc., San Diego, CA, USA) and sequenced on the NovaSeq 6000 platform (Illumina).
2.2. Mitochondrial Genome Assembly and Annotation
Before mitochondrial genome assembly, Trim_Galore (version 0.6.10) [8] was employed to remove reads containing low-quality bases and unknown bases (Ns) using the following options: “--quality 30 --length 121 --max_n 0” [9].
The assembly of the mitochondrial genome and annotation of protein-coding genes (PCGs) were performed using SPAdes (version 3.15.4) [10] and MitoZ (version 3.6) [11] with filtered reads. Moreover, MITOS1 [12] and MITOS2 [13] were utilized to verify the accuracy of the PCG annotation. The complete mitochondrial genome was visualized using Circos (version 0.69-8) [14].
2.3. Phylogenetic Analysis and Divergence Time Estimation
Two phylogenetic trees were reconstructed using datasets based on the cytochrome c oxidase subunit I gene (cox1) and 13 PCGs, respectively. For the cox1 dataset, 23 nucleotide sequences were obtained from the National Center for Biotechnology Information (NCBI): 21 from Cardiidae species and two from Pharidae species. MAFFT (version 7.475) [15] was employed to align these 23 nucleotide sequences. Subsequently, the best-fit nucleotide model for the cox1 dataset was identified using ModelFinder in IQ-TREE (version 2.2.0.3) [16]. For the 13 PCG dataset, only seven whole mitochondrial genomes from the Cardiidae family, including our data, were available at the NCBI (as of July 2024). Hence, nine mitochondrial genome sequences including two Pharidae species were used in this study. The nucleotide sequences of the 13 PCGs were aligned using MAFFT and concatenated. Next, PartitionFinder2 (version 2.1.1) [17] was used to determine the best-fit nucleotide model for each PCG based on the corrected Akaike information criterion (AICc).
Based on the cox1 and 13 PCG datasets, a maximum likelihood (ML) phylogenetic tree was reconstructed using RAxML-NG (version 0.9.0). To evaluate the robustness of the ML tree, 1000 bootstrapping iterations were performed. A Bayesian inference (BI) phylogenetic tree was also reconstructed using MrBayes (version 3.2.4) [18]. When generating the BI tree, two independent Markov chain Monte Carlo (MCMC) runs consisting of 1 × 107 generations were performed. To ensure convergence of the cold and heated chains, sampling was performed every 500 generations, and 25% of the samples were discarded as burn-in. FigTree (version 1.4.4) (http://tree.bio.ed.ac.uk/software/figtree, accessed on 1 July 2020) was employed to visualize the topology of the ML tree. Bootstrapping support (BS) based on the ML analysis and Bayesian posterior probability (BPP) based on the BI analysis were indicated at each node.
The divergence times of Cardiidae species were estimated using MCMCtree in PAML (version 4.10.5) [19]. The ML tree reconstructed using 13 PCGs was used as the input file. Moreover, three calibration points were utilized based on the TimeTree database [20]: Vasticardium flavum–Acanthocardia tuberculata (100.9–214.9 million years ago [MYA]), Hippopus porcellanus–Tridacna derasa (51.6 MYA), and the root age (210.1–514.5 MYA).
2.4. Positive Selection Analysis
To elucidate genomic response potentially associated with adaptation to environmental pressures, the CodeML program within the PAML package [19] was utilized to identify positively selected genes (PSGs) among the mitochondrial PCGs. The ML tree constructed using 13 mitochondrial genomes was utilized; the genomes were unrooted, and their branch lengths were removed (Figure 4a). Then, PAL2NAL (version 14) [21] was employed to align the nucleotide sequences.
A branch-site model was used to perform selection analysis. Using the branch-site model, a null model (model = 2, NSsites = 2, fix_omega = 1) was compared with an alternative model (model = 2, NSsites = 2, fix_omega = 0). This enabled the ratio of nonsynonymous (dN) to synonymous (dS) substitutions to vary among codon sites and the dN/dS ratio in the foreground branch to differ from that in the background branches. Likelihood ratio tests (LRTs) and chi-square (χ2) distributions were employed to determine the best-fit models and assess the statistical significance of the data (p values). If the LRT denoted statistical significance (p < 0.05) and the ω (dN/dS) ratio was greater than 1, the site was considered to be under positive selection.
AlphaFold2 (version 2.3.1) [22] was used to predict the three-dimensional (3D) structures of the 13 PCGs. PyMol (version 4.6) was employed for visualization [23].
3. Results and Discussions
3.1. General Information Regarding the Mitochondrial Genome of K. buelowi
In this study, 765,609,380 raw reads, equivalent to 116 Gb, were generated from the Keenocardium buelowi genome using Illumina paired-end sequencing. By following a stringent quality control process (Phred quality > 30), 653,573,610 clean reads were obtained, accounting for 85.37% of the original dataset (Table 1). These high-quality reads were then used to assemble the mitochondrial genome of K. buelowi.

Table 1.
General information on next-generation sequencing and mitochondrial genome assembly.
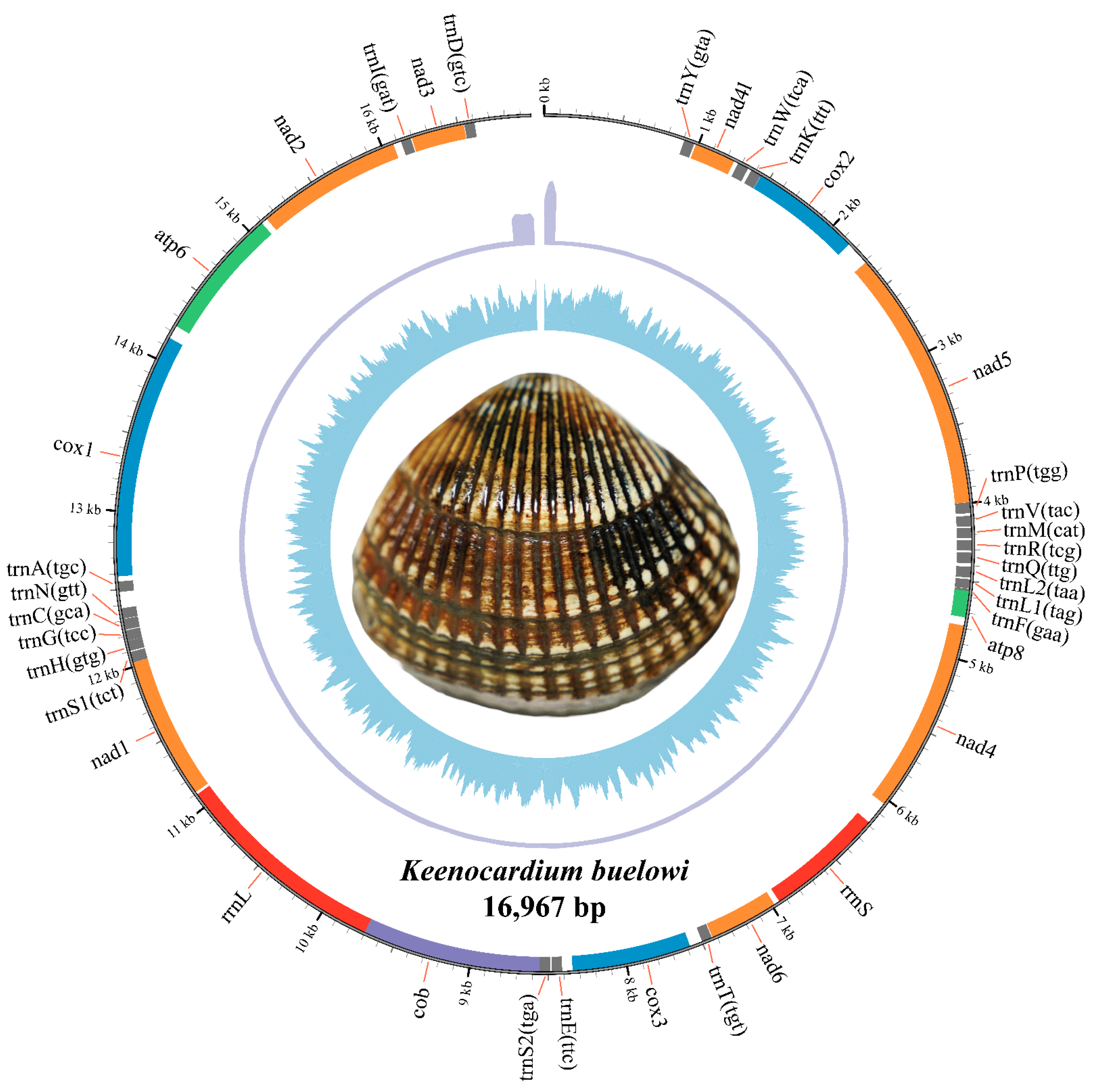
The complete mitochondrial genome of K. buelowi spanned 16,967 bp (Figure 1; GenBank: PP760258.1). The genome included 13 PCGs (atp6, atp8, cox1, cox2, cox3, cob, nad1, nad2, nad3, nad4, nad4l, nad5, and nad6), two ribosomal RNA (rRNA) genes (rrnS and rrnL), and 22 transfer RNA (tRNA) genes (trnY, trnW, trnK, trnP, trnV, trnM, trnR, trnQ, trnL2, trnL1, trnF, trnT, trnE, trnS2, trnS1, trnH, trnG, trnC, trnN, trnA, trnI, and trnD) (Table 2). Thirty-seven genes were located on the heavy strand and accounted for 88.25% of the entire mitochondrial genome. The 13 PCGs accounted for 64.71% of the entire mitochondrial genome. Most PCGs in K. buelowi began with either of the two start codons ATA (eight genes) or ATG (five genes). The stop codon employed in the mitochondrial genome was either TAG (seven genes) or TAA (six genes) (Table 2).
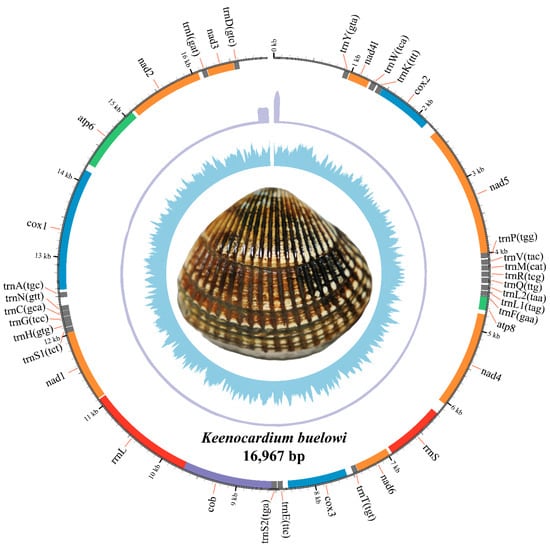
Figure 1.
The complete mitochondrial genome circular map of Keenocardium buelowi. The inner circle represents GC contents, the middle circle indicates sequencing depth, and the outer circle represents gene arrangements. Grey, orange, blue, green, red, and purple colors refer to trna, nad, cox, atp, rrna, and cob, respectively.
Table 2.
Organization of the Keenocardium buelowi mitochondrial genome.
3.2. Results of Phylogenetic Analysis
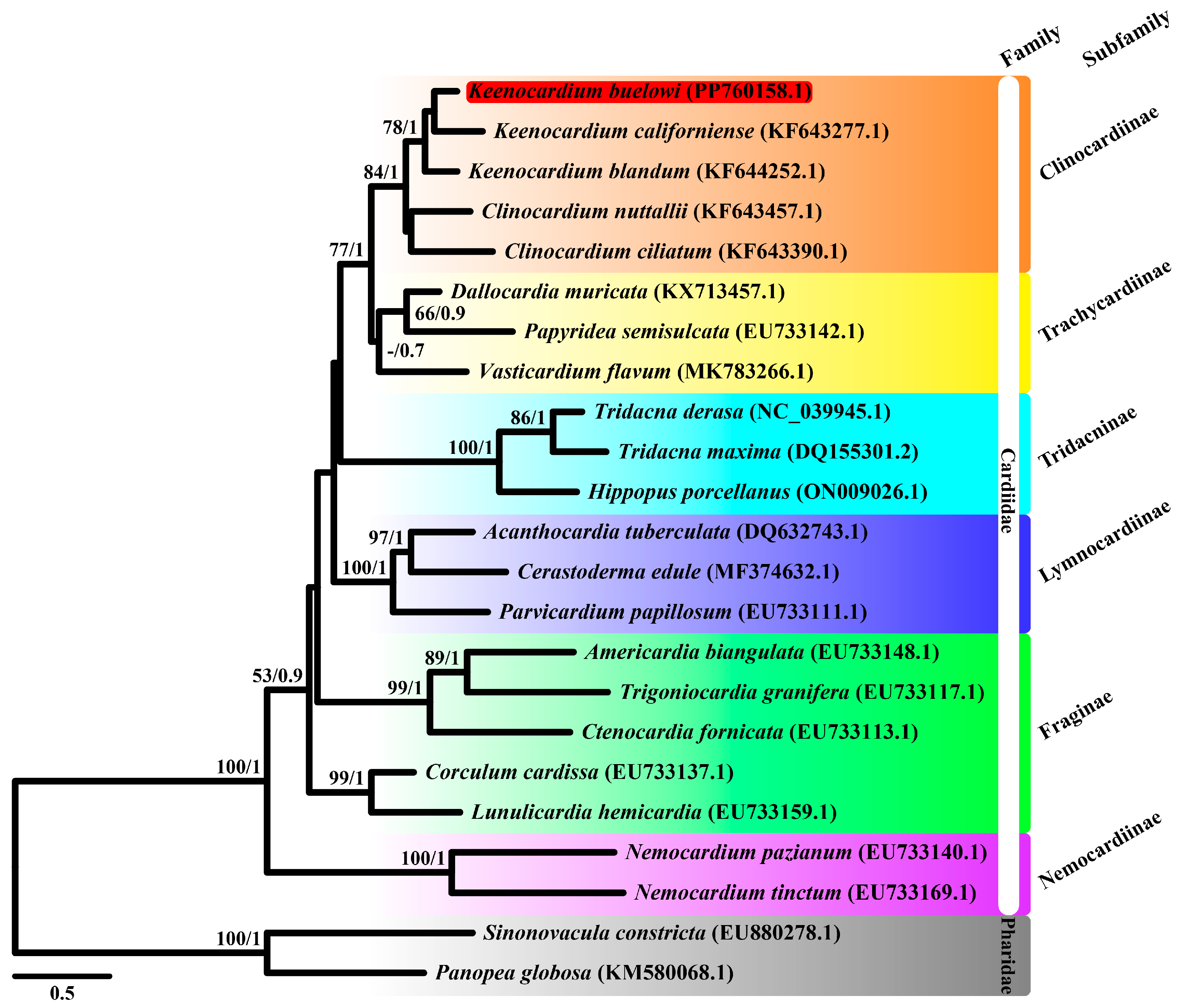
Although the construction of phylogenetic trees using multiple genes across various species is typically more informative than that using a single gene, we initially aimed to use 13 mitochondrial PCGs to elucidate phylogenetic relationships. However, due to the lack of complete mitochondrial genome sequences within the Clinocardiinae subfamily, cox1 sequences from 23 species were used instead (Figure 2). Moreover, a phylogenetic tree was reconstructed using 13 mitochondrial PCGs from nine species (Figure 3). Our study is important as it is the first to unravel the mitochondrial sequence within the Clinocardiinae subfamily, shedding light on taxonomic relationships at the subfamily level.
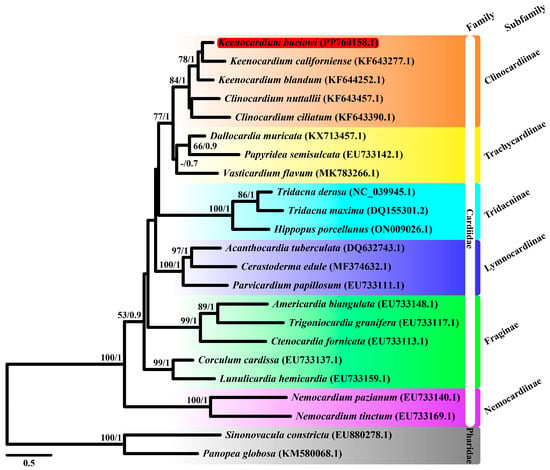
Figure 2.
Maximum likelihood (ML) tree of 21 Cardiidae species and two Pharidae species based on mitochondrial cytochrome c oxidase subunit I gene (cox1) sequences. Sequences from Sinonovacula constricta (EU880278.1) and Panopea globosa (KM580068.1) were used as outgroup species. Keenocardium buelowi was highlighted in red. The numbers on each node represent the bootstrap support (BS) from ML analysis and Bayesian posterior probability (BPP) from Bayesian inference analysis. Node supports with BS ≥ 60 and BPP ≥ 0.7 are indicated on each node. The scale bar represents the number of substitutions per site.
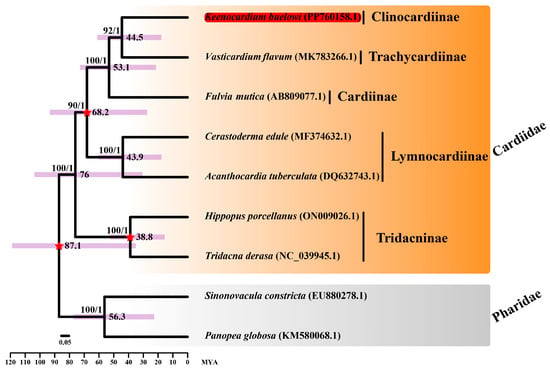
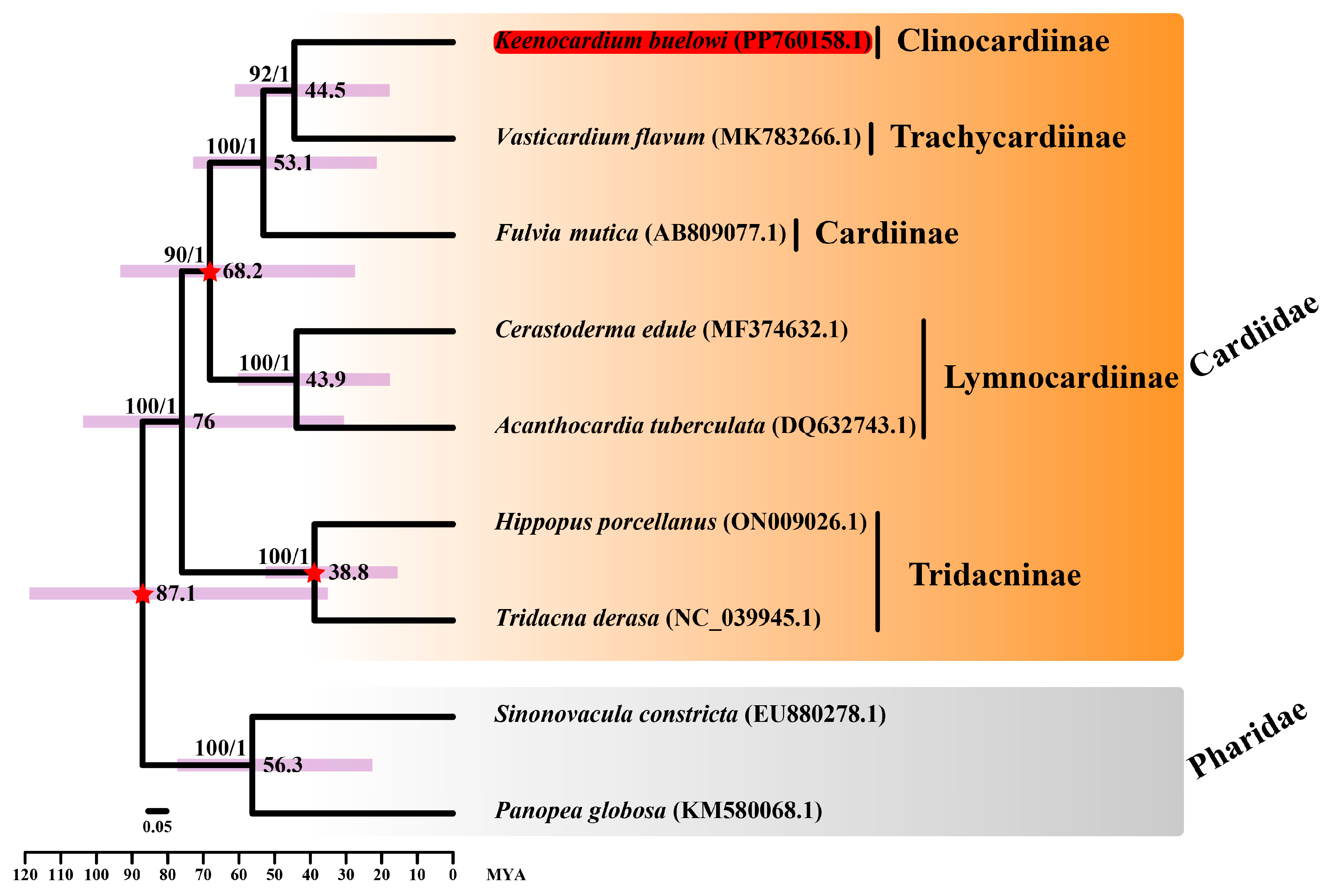
Figure 3.
Divergence time estimation analysis of seven Cardiidae species and two Pharidae species. Sequences from Sinonovacula constricta (EU880278.1) and Panopea globosa (KM580068.1) were used as outgroup species. Keenocardium buelowi was highlighted in red. The three calibration times used in this study are denoted with a red star in each node. The number on each node indicates the bootstrap support (BS) from ML analysis and Bayesian posterior probability (BPP) from Bayesian inference analysis. Node supports with BS ≥ 60 and BPP ≥ 0.7 are indicated on each node. Purple bars represent 95% highest posterior density (HPD) intervals, and the scale bar represents the number of substitutions per site.
ML and BI trees were constructed using two datasets: cox1 and 13 mitochondrial PCG datasets. For the cox1, the best-fit model was GTR + F + I + G4 according to the AICc. Since the atp8 gene was unavailable in three species (Acanthocardia tuberculata, Vasticardium flavum, and Sinnonovacula constricta), we manually reannotated it. For the 13 concatenated PCGs dataset, GTR + I + G was used for four genes (cox1, nad2, nad4l, and nad6), GTR + G was used for cox2, TVM + G was used for three genes (atp6, atp8, and cox3), and TVM + I + G was used for five genes (cob, nad1, nad3, nad4, and nad5). The ML and BI trees based on 13 PCGs showed identical topologies. From our study based on the cox1 dataset, Trachycardiinae was found to be the sister group to Clinocardiinae (BS = 77, BPP = 1) (Figure 2), consistent with the results based on the 13 PCG dataset (BS = 92, BPP = 1) (Figure 3). However, the relationships between Tridacninae and Lymnocardiinae differed in the cox1 and 13 PCG datasets (Figure 2 and Figure 3). The cox1 dataset exhibited weak node confidence (BS ≤ 32, BPP ≤ 0.5), whereas the 13 PCG dataset exhibited high node support (BS ≥ 90, BPP = 1) (Figure 2 and Figure 3). As trees based on multiple genes (13 genes in this study) generally provide stronger phylogenetic signals, we propose that Lymnocardiinae is the sister group to the Clinocardiinae + Trachycardiinae + Cardiinae clade. However, no complete mitochondrial data are currently available for Clinocardiinae. Moreover, only six complete mitochondrial sequences exist for Cardiidae. To better understand the phylogenetic positions of Cardiidae, additional mitochondrial genome sequences will be required.
The divergence time between K. buelowi and V. flavum was estimated to be 44.5 MYA (95% CI: 18–60.9) (Figure 3). Moreover, the divergence time between Cerastoderma edule and A. tuberculata was estimated to be 43.9 MYA (95% CI: 17.8–60.2), consistent with the findings of Li et al. [24]. The divergence time between V. flavum and F. mutica overlapped with the estimates provided by Herrera et al. [25]. However, the rapid substitution rates in the mitochondrial genome hinder the precise estimation of deep divergence times [26]. Given the influence of horizontal gene transfer, frequent introgression, and incomplete lineage sorting, phylogenetic research solely based on mitochondrial sequences is inherently limited [27,28]. Therefore, nuclear and mitochondrial sequences may provide contrasting or complementary information regarding tree topologies and branch lengths [29,30].
3.3. Selection Patterns in Mitochondrial Genes of K. buelowi
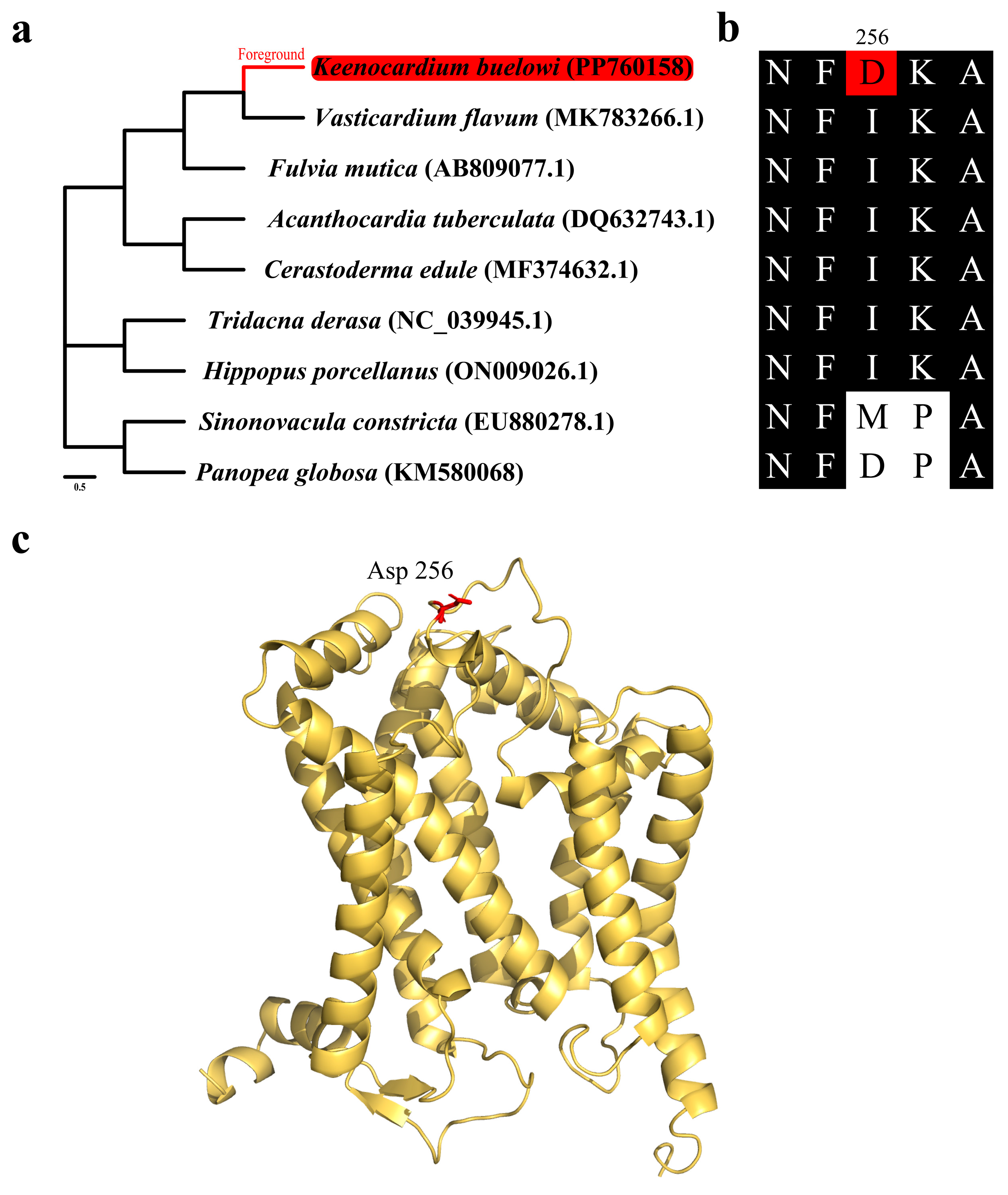
Mitochondrial genes play a vital role in cellular function, and as a result, purifying selection is the primary force shaping their evolution. Therefore, identifying signatures of positive selection provide valuable insights into how these genes may contribute to adaptive responses to the environment pressures. To identify genes subjected to environmental pressures, the ω ratio (dN/dS) was calculated for each of the 13 mitochondrial PCGs and compared (Figure 4 and Table 2). The ω ratio was used to determine the natural selection patterns for these PCGs [31]. The branch-site model revealed that all PCGs, except for cob, exhibited signals of purifying selection during evolution (Table 3). However, the results of cob were significant in the LRT (LR = 6.97, p < 0.008), suggesting that it underwent strong selection pressure throughout its evolutionary history. Specifically, residue 256 in six Cardiidae species was isoleucine, a hydrophobic amino acid, which was substituted by aspartic acid, a negatively charged amino acid, in K. buelowi. This alteration suggests a potential adaptive response. However, the functional properties of these substitutions still need to be elucidated through further physiological experiments. Jacobsen et al. reported that mutations in mitochondrial genes are linked to their positions within the mitochondrial genome [32]. This aligns with our findings that cob may have evolved more rapidly because of being distant from the origin of mitochondrial genome replication (Figure 1 and Table 3) [33]. As protein structures and functions depend on amino acid interactions, the placement of Asp256 within the 3D structure was further examined in our study (Figure 4b). The positively selected residue was located close to the periplasmic membrane region (Figure 4c). Moreover, cob exhibited strong positive selection pressure, suggesting that cob plays a crucial role in the evolution of the K. buelowi mitochondrial genome. However, the correlation between positive selection pressure on cob and environmental variables was limited in our study. Nonetheless, this result may be influenced by other variables not evaluated in our study.
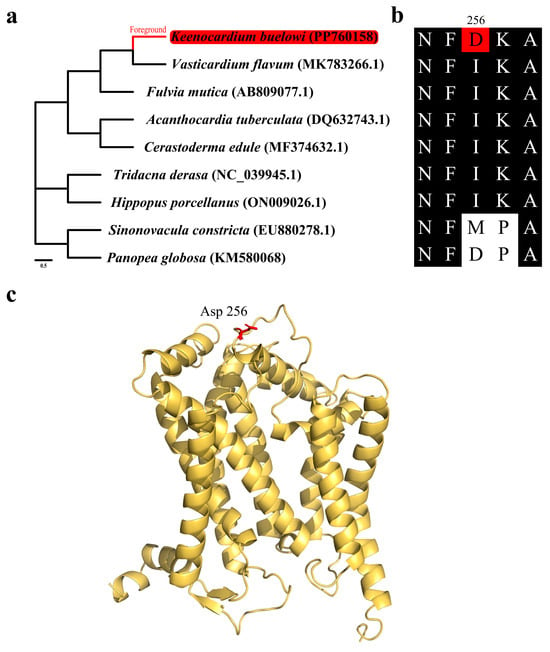
Figure 4.
Positive selection analysis results of the mitochondrial cytochrome b gene (cob). (a) Species tree used for positive selection analysis. The Keenocardium buelowi branch was used as a foreground. (b) Multiple sequence alignment (MSA) of mitochondrial cob from nine bivalves. Within the alignment, the red background represents mutated residues (Asp256), while the black background represents highly conserved regions. (c) Yellow ribbon diagram showing the three-dimensional structure of mitochondrial cob. The red residue (Asp256) indicates a positively selected site from the branch-site model.
Table 3.
Results of positive selection analysis using branch-site model. Abbreviations: np (number of parameters), LRT (likelihood ratio test), BEB (Bayes empirical Bayes).
4. Conclusions
In this study, next-generation sequencing was performed to assemble and annotate the 16,967-bp mitochondrial genome of Keenocardium buelowi, providing the first complete mitochondrial genome within the Clinocardiinae subfamily. Our comprehensive analysis assessed the molecular characteristics of the K. buelowi mitochondrial genome. ML and BI trees were reconstructed based on cox1 and 13 PCG datasets. Our findings indicated that Clinocardiinae forms a monophyletic group comprising two genera: Keenocardium and Clinocardium. Moreover, our data confirmed that Clinocardiinae is the sister group to Trachycardiinae. The divergence times for seven Cardiidae species were estimated using 13 mitochondrial PCGs. Furthermore, our results revealed that cob shows signs of selection pressure, suggesting its significant role in the evolution of K. buelowi. Further research is needed to improve the resolution of phylogenetic relationships among Cardiidae species since complete mitochondrial genome for this family is still lacking.
Author Contributions
Conceptualization, S.-i.E.; methodology, H.C., Y.G., Y.K.A. and S.-i.E.; formal analysis, H.C.; data curation, H.C. and Y.G.; writing—original draft preparation, H.C.; writing—review and editing, H.C., Y.G., Y.K.A. and S.-i.E.; and project administration, Y.K.A. and S.-i.E. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (2022R1A2C4002058), and Korea Institute of Marine Science & Technology Promotion (RS-2022-KS221676) funded by the Ministry of Oceans and Fisheries.
Institutional Review Board Statement
Not applicable. Ethics approval is not necessary for the research of Invertebrates.
Informed Consent Statement
Not applicable.
Data Availability Statement
The complete mitochondrial genome sequence of Keenocardium buelowi is available in GenBank under the accession number PP760158.1.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Ahyong, S.; Boyko, C.B.; Bily, N.; Bernot, J.; Bieler, R.; Brandao, S.N.; Daly, M.; De Grave, S.; Gofas, S.; Hernandez, F.; et al. World Register of Marine Species (WoRMS); Flanders Marine Institute: Ostend, Belgium, 2023. [Google Scholar]
- Bao, Y.; Zeng, Q.; Wang, J.; Zhang, Z.; Zhang, Y.; Wang, S.; Wong, N.K.; Yuan, W.; Huang, Y.; Zhang, W.; et al. Genomic insights into the origin and evolution of Molluscan red-bloodedness in the blood clam Tegillarca granosa. Mol. Biol. Evol. 2021, 38, 2351–2365. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, H.M.; Cho, Y.G.; Shin, J.S.; Yoo, J.W.; Hong, H.K.; Choi, K.S. Effects of spawning stress on the immune capacity of blood cockle Tegillarca granosa occurring on the south coast of Korea. Fish Shellfish Immunol. 2022, 120, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.S.; Kang, D.H.; Park, H.S.; Choi, K.S. Seasonal changes in reproduction and biochemical composition of the cockle, Fulvia mutica reeve (1884), in Cheonsu Bay off the West Coast of Korea. J. Shellfish Res. 2011, 30, 95–101. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Choi, Y.J.; Rohmah, Z.; Jeong, S.B.; Hwang, D.J.; Jung, Y.G.; Choi, B.D. Seasonal variations of nutritional components in cockles (Tegillarca granosa) processed from the Southern Coast of Korea. Cogent Food Agric. 2017, 3, 1360102. [Google Scholar] [CrossRef]
- Javonillo, R.; Malabarba, L.R.; Weitzman, S.H.; Burns, J.R. Relationships among major lineages of characid fishes (Teleostei: Ostariophysi: Characiformes), based on molecular sequence data. Mol. Phylogenet. Evol. 2010, 54, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Hiesel, R.; von Haeseler, A.; Brennicke, A. Plant mitochondrial nucleic acid sequences as a tool for phylogenetic analysis. Proc. Natl. Acad. Sci. USA 1994, 91, 634–638. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Choi, H.; Kim, S.L.; Jeong, M.K.; Yu, O.H.; Eyun, S. Identification and phylogenetic analysis of chitin synthase genes from the deep-sea polychaete Branchipolynoe onnuriensis genome. J. Mar. Sci. Eng. 2022, 10, 598. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Donath, A.; Jühling, F.; Al-Arab, M.; Bernhart, S.H.; Reinhardt, F.; Stadler, P.F.; Middendorf, M.; Bernt, M. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 2019, 47, 10543–10552. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL. Version: 2.4.0. 2020. Available online: http://www.pymol.org/pymol (accessed on 25 August 2024).
- Li, J.; Lemer, S.; Kirkendale, L.; Bieler, R.; Cavanaugh, C.M.; Giribet, G. Shedding light: A phylotranscriptomic perspective illuminates the origin of photosymbiosis in marine bivalves. BMC Evol. Biol. 2020, 20, 50. [Google Scholar] [CrossRef]
- Herrera, N.D.; ter Poorten, J.J.; Bieler, R.; Mikkelsen, P.M.; Strong, E.E.; Jablonski, D.; Steppan, S.J. Molecular phylogenetics and historical biogeography amid shifting continents in the cockles and giant clams (Bivalvia: Cardiidae). Mol. Phylogenet. Evol. 2015, 93, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Zardoya, R.; Meyer, A. Phylogenetic performance of mitochondrial protein-coding genes in resolving relationships among vertebrates. Mol. Biol. Evol. 1996, 13, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.W.O.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.M.A.; Levin, S.A. Leaky prezygotic isolation and porous genomes: Rapid introgression of maternally inherited DNA. Evolution 2005, 59, 720–729. [Google Scholar] [CrossRef]
- Salzburger, W.; Meyer, A.; Baric, S.; Verheyen, E.; Sturmbauer, C. Phylogeny of the lake Tanganyika cichlid species flock and its relationship to the central and East African haplochromine cichlid fish faunas. Syst. Biol. 2002, 51, 113–135. [Google Scholar] [CrossRef]
- Sturmbauer, C.; Salzburger, W.; Duftner, N.; Schelly, R.; Koblmüller, S. Evolutionary history of the lake Tanganyika cichlid tribe Lamprologini (Teleostei: Perciformes) derived from mitochondrial and nuclear DNA data. Mol. Phylogenet. Evol. 2010, 57, 266–284. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486. [Google Scholar] [CrossRef]
- Jacobsen, M.W.; da Fonseca, R.R.; Bernatchez, L.; Hansen, M.M. Comparative analysis of complete mitochondrial genomes suggests that relaxed purifying selection is driving high nonsynonymous evolutionary rate of the NADH2 gene in whitefish (Coregonus ssp.). Mol. Phylogenet. Evol. 2016, 95, 161–170. [Google Scholar] [CrossRef]
- Zhao, D.; Guo, Y.; Gao, Y. Natural selection drives the evolution of mitogenomes in Acrossocheilus. PLoS ONE 2022, 17, e0276056. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).