
Genome-Wide Scan for Copy Number Variations in Chinese Merino Sheep Based on Ovine High-Density 600K SNP Arrays
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection and Genotyping
2.3. Data Quality Control and CNV Calling
2.4. qPCR Confirmation of CNVRs
2.5. Functional Enrichment Analysis Procedures for CNVRs
3. Results
3.1. CNV Genotyping
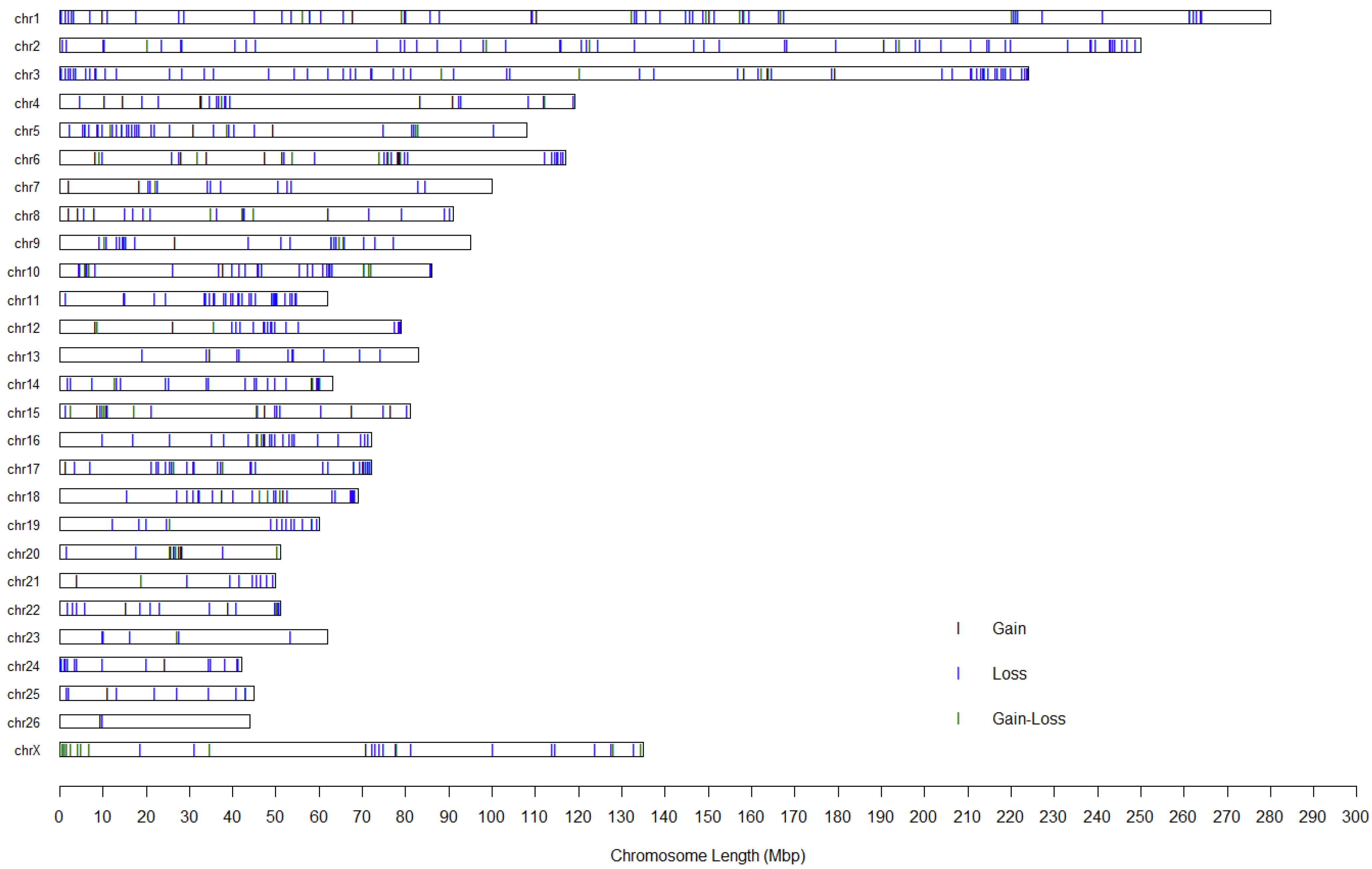
3.2. Genome-Wide Surveys of CNVs and CNVRs in Fine Wool Sheep
3.3. CNV Validation by Quantitative PCR
3.4. Functional Enrichment Analysis of CNVRs
4. Discussion

{kind=link}
{kind=link}
{kind=link}
| Findings from Different Studies | CNVRs Overlapping with This Study | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Platform | Study | Species | Sample | Count | Total Length (Mbs) | Loss | Gain | Both | Count | Percentage of Count | Total Length (Mbs) | Percentage Length |
| CGH | Jenkins et al. (2016) [73] | Texel, Coopworth, Perendale, | 30 | 3488 | 67.6 | 2023 | 1325 | 140 | 65 | 9.90% | 5.82 | 13.20% |
| Romney, and Merino | ||||||||||||
| Fontanesi et al. (2011) [75] | Bagnolese, Comisana, Laticauda, Massese, Sarda, and Valle del Belice | 11 | 135 | 10.5 | 75 | 59 | 1 | 0 | 0% | 0 | 0% | |
| Hou et al. (2015) [74] | Mongolian sheep, Kazakh | 122 | 51 | 15.55 | 21 | 23 | 7 | 1 | 0.15% | 0.05 | ||
| Sheep, Tibetan sheep, Hu sheep | ||||||||||||
| SNP50 | Yang et al. (2018) [65] | Sixty-eight breeds from Africa, America, Asia, and Europe | 2254 | 619 | 197 | - | - | - | 66 | 10.06% | 17.85 | 40.66% |
| Di Gerlando et al. (2019) [66] | Valle del Belice sheep | 468 | 365 | 118.36 | 320 | 43 | 2 | |||||
| Goyache et al. (2021) [76] | Djallonké (West African Dwarf) sheep | 184 | 63 | 82.5 | 36 | 7 | 20 | |||||
| Taghizadeh et al. (2022) [67] | Baluchi sheep Lori-Bakhtiari sheep Zel sheep | 192 | 515 | 73.85 | 141 | 364 | 10 | |||||
| Ma et al. (2015) [77] | Dorper, Poll Dorset, Texel, Suffolk, South American Mutton Merino, Borderdale, Gansu Alpine Merino, and Gansu Morden sheep | 160 | 111 | 13.75 | 12 | 99 | 0 | 3 | 0.45% | 0.22 | 0.50% | |
| Liu et al. (2013) [72] | German Mutton, Dorper, and Sunite | 100 | 238 | 60.35 | 219 | 13 | 6 | 15 | 2.20% | 1.69 | 3.80% | |
| SNP600 | Ma et al. (2017) [68] | Chinese Tan sheep | 48 | 1296 | 121.8 | 1173 | 119 | 5 | 184 | 28.00% | 24.4 | 55.60% |
| Zhu et al. (2016) [24] | Chinese indigenous sheep | 120 | 490 | 81.04 | 390 | 93 | 7 | 165 | 25.10% | 9.31 | 21.20% | |
| Wang et al. (2020) [69] | Hu sheep DS sheep SHH sheep | 270 | 919 | 48.17 | 730 | 102 | 87 | |||||
| Salehian-Dehkordi et al. (2021) [70] | sheep from 67 populations all over the world | 2059 | 1217 | 245 | 918 | 197 | 102 | |||||
| This Study | Chinese Merino sheep | 288 | 656 | 43.9 | 519 | 60 | 77 | - | - | - | - | |
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanchez, S.; Juarez, U.; Dominguez, J.; Molina, B.; Barrientos, R.; Martinez-Hernandez, A.; Carnevale, A.; Grether-Gonzalez, P.; Mayen, D.G.; Villarroel, C.; et al. Frequent copy number variants in a cohort of Mexican-Mestizo individuals. Mol. Cytogenet. 2023, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Gordeeva, V.; Sharova, E.; Arapidi, G. Progress in Methods for Copy Number Variation Profiling. Int. J. Mol. Sci. 2022, 23, 2143. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Singh, S.; Nandhini, P.B.; Bhatia, A.K.; Dixit, S.P.; Ganguly, I. Comparative genomic diversity analysis of copy number variations (CNV) in indicine and taurine cattle thriving in Europe and Indian subcontinent. Anim. Biotechnol. 2023, 34, 3483–3494. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Bao, Q.; Ma, X.; Chu, M.; Huang, C.; Guo, X.; Liang, C.; Yan, P. Analysis of copy number variation in the whole genome of normal-haired and long-haired tianzhu white yaks. Genes 2022, 13, 2405. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, Y.; Liu, S.; Meng, Q. Genome-Wide Assessment Characteristics of Genes Overlapping Copy Number Variation Regions in Duroc Purebred Population. Front. Genet. 2021, 12, 753748. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, W.; Jiang, Y.; Zhou, M.; Liu, L.; Su, S.; Li, X.; Wang, C. Genome-Wide Detection and Analysis of Copy Number Variation in Anhui Indigenous and Western Commercial Pig Breeds Using Porcine 80K SNP BeadChip. Genes 2023, 14, 654. [Google Scholar] [CrossRef]
- Zamariolli, M.; Auwerx, C.; Sadler, M.C.; van der Graaf, A.; Lepik, K.; Schoeler, T.; Moyses-Oliveira, M.; Dantas, A.G.; Melaragno, M.I.; Kutalik, Z. The impact of 22q11.2 copy-number variants on human traits in the general population. Am. J. Hum. Genet. 2023, 110, 300–313. [Google Scholar] [CrossRef]
- Benedetti, F.; Silvestri, G.; Saadat, S.; Denaro, F.; Latinovic, O.S.; Davis, H.; Williams, S.; Bryant, J.; Ippodrino, R.; Rathinam, C.V.; et al. Mycoplasma DnaK increases DNA copy number variants in vivo. Proc. Natl. Acad. Sci. USA 2023, 120, e2219897120. [Google Scholar] [CrossRef]
- Liu, G.; Yang, H.; Yuan, X. A shortest path-based approach for copy number variation detection from next-generation sequencing data. Front. Genet. 2022, 13, 1084974. [Google Scholar] [CrossRef]
- Mihola, O.; Kobets, T.; Krivankova, K.; Linhartova, E.; Gasic, S.; Schimenti, J.C.; Trachtulec, Z. Copy-number variation introduced by long transgenes compromises mouse male fertility independently of pachytene checkpoints. Chromosoma 2020, 129, 69–82. [Google Scholar] [CrossRef]
- Ueo, D.; Furuhashi, M.; Sasaki, T.; Kudoh, J.; Parry, D.A.; Winter, D.J.; Sasaki, T.; Hashimoto, T.; Tsuruta, D.; Fujiwara, S. Intragenic copy number variation in mouse epiplakin 1 (Eppk1) and the conservation of the repeat structures in the lower vertebrates. J. Dermatol. Sci. 2021, 103, 186–189. [Google Scholar] [CrossRef] [PubMed]
- White, J.; O’Neill, M.; Sheth, K.; Lamb, D. MP65-12 Murine Rbfox-2 Haploinsufficiency Parallels Congenital Anomalies in Human Patients with Rbfox-2 Copy Number Variants. J. Urol. 2020, 203, e978. [Google Scholar] [CrossRef]
- Keshavarz, M.; Savriama, Y.; Refki, P.; Reeves, R.G.; Tautz, D. Natural copy number variation of tandemly repeated regulatory SNORD RNAs leads to individual phenotypic differences in mice. Mol. Ecol. 2021, 30, 4708–4722. [Google Scholar] [CrossRef] [PubMed]
- Serres-Armero, A.; Davis, B.W.; Povolotskaya, I.S.; Morcillo-Suarez, C.; Plassais, J.; Juan, D.; Ostrander, E.A.; Marques-Bonet, T. Copy number variation underlies complex phenotypes in domestic dog breeds and other canids. Genome Res. 2021, 31, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Cerezo-Echevarria, A.; Kehl, A.; Beitzinger, C.; Müller, T.; Klopfleisch, R.; Aupperle-Lellbach, H. Evaluating the histologic grade of digital squamous cell carcinomas in dogs and copy number variation of KIT Ligand—A correlation study. Vet. Sci. 2023, 10, 88. [Google Scholar] [CrossRef]
- Di Gerlando, R.; Mastrangelo, S.; Sardina, M.T.; Ragatzu, M.; Spaterna, A.; Portolano, B.; Biscarini, F.; Ciampolini, R. A genome-wide detection of copy number variations using SNP genotyping arrays in braque français type pyrénées dogs. Animals 2019, 9, 77. [Google Scholar] [CrossRef]
- Rafter, P.; Gormley, I.C.; Parnell, A.C.; Naderi, S.; Berry, D.P. The Contribution of Copy Number Variants and Single Nucleotide Polymorphisms to the Additive Genetic Variance of Carcass Traits in Cattle. Front. Genet. 2021, 12, 761503. [Google Scholar] [CrossRef]
- Buggiotti, L.; Yudin, N.S.; Larkin, D.M. Copy number variants in two northernmost cattle breeds are related to their adaptive phenotypes. Genes 2022, 13, 1595. [Google Scholar] [CrossRef]
- Kumar, H.; Panigrahi, M.; Saravanan, K.; Rajawat, D.; Parida, S.; Bhushan, B.; Gaur, G.; Dutt, T.; Mishra, B.; Singh, R. Genome-wide detection of copy number variations in Tharparkar cattle. Anim. Biotechnol. 2023, 34, 448–455. [Google Scholar] [CrossRef]
- Wu, Y.; Fan, H.; Jing, S.; Xia, J.; Chen, Y.; Zhang, L.; Gao, X.; Li, J.; Gao, H.; Ren, H. A genome-wide scan for copy number variations using high-density single nucleotide polymorphism array in Simmental cattle. Anim. Genet. 2015, 46, 289–298. [Google Scholar] [CrossRef]
- Yi, G.; Qu, L.; Liu, J.; Yan, Y.; Xu, G.; Yang, N. Genome-wide patterns of copy number variation in the diversified chicken genomes using next-generation sequencing. BMC Genom. 2014, 15, 962. [Google Scholar] [CrossRef] [PubMed]
- Keel, B.N.; Nonneman, D.J.; Lindholm-Perry, A.K.; Oliver, W.T.; Rohrer, G.A. A survey of copy number variation in the porcine genome detected from whole-genome sequence. Front. Genet. 2019, 10, 737. [Google Scholar] [CrossRef] [PubMed]
- Nandolo, W.; Mészáros, G.; Wurzinger, M.; Banda, L.J.; Gondwe, T.N.; Mulindwa, H.A.; Nakimbugwe, H.N.; Clark, E.L.; Woodward-Greene, M.J.; Liu, M. Detection of copy number variants in African goats using whole genome sequence data. BMC Genom. 2021, 22, 398. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Fan, H.; Yuan, Z.; Hu, S.; Ma, X.; Xuan, J.; Wang, H.; Zhang, L.; Wei, C.; Zhang, Q. Genome-wide detection of CNVs in Chinese indigenous sheep with different types of tails using ovine high-density 600K SNP arrays. Sci. Rep. 2016, 6, 27822. [Google Scholar] [CrossRef]
- Saitou, M.; Gokcumen, O. An evolutionary perspective on the impact of genomic copy number variation on human health. J. Mol. Evol. 2020, 88, 104–119. [Google Scholar] [CrossRef]
- Isa, I.I.M.; Jamaluddin, J.; Achim, N.H.; Abubakar, S. Population-specific profiling of CCL3L1 copy number of the three major ethnic groups in Malaysia and the implication on HIV susceptibility. Gene 2020, 754, 144821. [Google Scholar]
- Qi, Y.; Zhou, X.; Bu, D.; Hou, P.; Lv, J.; Zhang, H. Low copy numbers of FCGR3A and FCGR3B associated with Chinese patients with SLE and AASV. Lupus 2017, 26, 1383–1389. [Google Scholar] [CrossRef]
- Liang, X.; Lan, J.; Xu, M.; Qin, K.; Liu, H.; Sun, G.; Liu, X.; Chen, Y.; He, Z. Impact of KIT editing on Coat pigmentation and Fresh Meat Color in Yorkshire pigs. CRISPR J. 2022, 5, 825–842. [Google Scholar] [CrossRef]
- Nowacka-Woszuk, J.; Mackowski, M.; Stefaniuk-Szmukier, M.; Cieslak, J. The equine graying with age mutation of the STX17 gene: A copy number study using droplet digital PCR reveals a new pattern. Anim. Genet. 2021, 52, 223–227. [Google Scholar] [CrossRef]
- Feng, Z.; Li, X.; Cheng, J.; Jiang, R.; Huang, R.; Wang, D.; Huang, Y.; Pi, L.; Hu, L.; Chen, H. Copy number variation of the PIGY gene in sheep and its association analysis with growth traits. Animals 2020, 10, 688. [Google Scholar] [CrossRef]
- Henriksen, R.A.; Zhao, L.; Korneliussen, T.S. NGSNGS: Next-generation simulator for next-generation sequencing data. Bioinformatics 2023, 39, btad041. [Google Scholar] [CrossRef] [PubMed]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; Von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Song, C. Responses of Chinese merino sheep (Junken Type) on copper-deprived natural pasture. Biol. Trace Elem. Res. 2021, 199, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.W.; Bi, W. Novel applications of array comparative genomic hybridization in molecular diagnostics. Expert Rev. Mol. Diagn. 2018, 18, 531–542. [Google Scholar] [CrossRef]
- Zhan, B.; Fadista, J.; Thomsen, B.; Hedegaard, J.; Panitz, F.; Bendixen, C. Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping. BMC Genom. 2011, 12, 557. [Google Scholar] [CrossRef]
- Sastre, N.; Mercadé, A.; Casellas, J. SNP+ to predict dropout rates in SNP arrays. Conserv. Genet. Resour. 2023, 15, 113–116. [Google Scholar] [CrossRef]
- Ceccobelli, S.; Landi, V.; Senczuk, G.; Mastrangelo, S.; Sardina, M.T.; Ben-Jemaa, S.; Persichilli, C.; Karsli, T.; Bâlteanu, V.-A.; Raschia, M.A. A comprehensive analysis of the genetic diversity and environmental adaptability in worldwide Merino and Merino-derived sheep breeds. Genet. Sel. Evol. 2023, 55, 24. [Google Scholar] [CrossRef]
- Li, X.; Yang, J.; Shen, M.; Xie, X.-L.; Liu, G.-J.; Xu, Y.-X.; Lv, F.-H.; Yang, H.; Yang, Y.-L.; Liu, C.-B. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef]
- Mu, F.; Rong, E.; Jing, Y.; Yang, H.; Ma, G.; Yan, X.; Wang, Z.; Li, Y.; Li, H.; Wang, N. Structural characterization and association of ovine Dickkopf-1 gene with wool production and quality traits in Chinese Merino. Genes 2017, 8, 400. [Google Scholar] [CrossRef]
- Roldan, D.; Dodero, A.; Bidinost, F.; Taddeo, H.; Allain, D.; Poli, M.; Elsen, J.M. Merino sheep: A further look at quantitative trait loci for wool production. Animal 2010, 4, 1330–1340. [Google Scholar] [CrossRef]
- Li, S.; Xi, Q.; Zhao, F.; Wang, J.; He, Z.; Hu, J.; Liu, X.; Luo, Y. A highly polymorphic caprine keratin-associated protein gene identified and its effect on cashmere traits. J. Anim. Sci. 2021, 99, skab233. [Google Scholar] [CrossRef] [PubMed]
- Powell, B.a.; Rogers, G. The role of keratin proteins and their genes in the growth, structure and properties of hair. Exs 1997, 78, 59–148. [Google Scholar] [PubMed]
- Parsons, Y.; Piper, L.; Cooper, D. Linkage relationships between keratin-associated protein (KRTAP) genes and growth hormone in sheep. Genomics 1994, 20, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Zhou, H.; Forrest, R.H.; Li, S.; Wang, J.; Dyer, J.M.; Luo, Y.; Hickford, J.G. Wool keratin-associated protein genes in sheep—A review. Genes 2016, 7, 24. [Google Scholar] [CrossRef]
- Zhou, H.; Gong, H.; Li, S.; Luo, Y.; Hickford, J. A 57-bp deletion in the ovine KAP 6-1 gene affects wool fibre diameter. J. Anim. Breed. Genet. 2015, 132, 301–307. [Google Scholar] [CrossRef]
- Gong, H.; Zhou, H.; Hodge, S.; Dyer, J.M.; Hickford, J.G. Association of wool traits with variation in the ovine KAP1-2 gene in Merino cross lambs. Small Rumin. Res. 2015, 124, 24–29. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Li, W.; Tao, J.; Hickford, J.G. Exploring variation in ovine KRTAP19-5 and its effect on fine wool fibre curvature in Chinese Tan sheep. Animals 2024, 14, 2155. [Google Scholar] [CrossRef]
- Chai, W.; Zhou, H.; Gong, H.; Wang, J.; Luo, Y.; Hickford, J. Nucleotide variation in the ovine KRT31 promoter region and its association with variation in wool traits in Merino-cross lambs. J. Agric. Sci. 2019, 157, 182–188. [Google Scholar] [CrossRef]
- Chai, W.; Zhou, H.; Gong, H.; Hickford, J.G. Variation in the ovine KRT34 promoter region affects wool traits. Small Rumin. Res. 2022, 206, 106586. [Google Scholar] [CrossRef]
- Li, W.; Bai, L.; Zhou, H.; Zhang, Z.; Ma, Z.; Wu, G.; Luo, Y.; Tanner, J.; Hickford, J.G. Ovine KRT81 Variants and Their Influence on Selected Wool Traits of Commercial Value. Genes 2024, 15, 681. [Google Scholar] [CrossRef]
- Yu, X.; Li, S.; Zhou, H.; Zhao, F.; Hu, J.; Wang, J.; Liu, X.; Li, M.; Zhao, Z.; Hao, Z. Spatiotemporal expression and haplotypes identification of KRT84 gene and their association with wool traits in Gansu Alpine fine-wool sheep. Genes 2024, 15, 248. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Ha, S.-J.; Seong, H.-S.; Choi, J.-Y.; Baek, H.-J.; Yang, B.-C.; Choi, J.-W.; Kim, N.-Y. Identification of copy number variations in four horse breed populations in south korea. Animals 2022, 12, 3501. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.H.; Yang, F.; Marques-Bonet, T.; Murphy, C.; Fitzgerald, T.; Lee, A.S.; Hyland, C.; Stone, A.C.; Hurles, M.E.; Tyler-Smith, C. Copy number variation and evolution in humans and chimpanzees. Genome Res. 2008, 18, 1698–1710. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Di Gerlando, R.; Mastrangelo, S.; Tolone, M.; Rizzuto, I.; Sutera, A.M.; Moscarelli, A.; Portolano, B.; Sardina, M.T. Identification of copy number variations and genetic diversity in Italian insular sheep breeds. Animals 2022, 12, 217. [Google Scholar] [CrossRef]
- Molparia, B.; Oliveira, G.; Wagner, J.L.; Spencer, E.G.; Torkamani, A. A feasibility study of colorectal cancer diagnosis via circulating tumor DNA derived CNV detection. PLoS ONE 2018, 13, e0196826. [Google Scholar] [CrossRef]
- Zou, J.; Wang, E. eTumorType, An algorithm of discriminating cancer types for circulating tumor cells or cell-free DNAs in blood. Genom. Proteom. Bioinform. 2017, 15, 130–140. [Google Scholar] [CrossRef]
- Huang, J.; Li, R.; Zhang, X.; Huang, Y.; Dang, R.; Lan, X.; Chen, H.; Lei, C. Copy number veriation regions detection in Qinchuan cattle. Livest. Sci. 2017, 204, 88–91. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, J.; Guo, Y.; Xu, Q.; Liu, M.; Cheng, M.; Chao, X.; Schinckel, A.P.; Zhou, B. Genome-wide detection of copy number variations and evaluation of candidate copy number polymorphism genes associated with complex traits of pigs. Front. Vet. Sci. 2022, 9, 909039. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhang, J.; Zhang, C.; Shi, Y.; Zhao, B.; Jiao, A.; Chen, B. Using Illumina Infinium HumanMethylation 450K BeadChip to explore genome-wide DNA methylation profiles in a human hepatocellular carcinoma cell line. Mol. Med. Rep. 2018, 18, 4446–4456. [Google Scholar] [CrossRef] [PubMed]
- Lepamets, M.; Auwerx, C.; Nõukas, M.; Claringbould, A.; Porcu, E.; Kals, M.; Jürgenson, T.; Morris, A.P.; Võsa, U.; Bochud, M. Omics-informed CNV calls reduce false-positive rates and improve power for CNV-trait associations. Hum. Genet. Genom. Adv. 2022, 3, 100133. [Google Scholar] [CrossRef]
- Yang, L.; Xu, L.; Zhou, Y.; Liu, M.; Wang, L.; Kijas, J.W.; Zhang, H.; Li, L.; Liu, G.E. Diversity of copy number variation in a worldwide population of sheep. Genomics 2018, 110, 143–148. [Google Scholar] [CrossRef]
- Di Gerlando, R.; Sutera, A.M.; Mastrangelo, S.; Tolone, M.; Portolano, B.; Sottile, G.; Bagnato, A.; Strillacci, M.G.; Sardina, M.T. Genome-wide association study between CNVs and milk production traits in Valle del Belice sheep. PLoS ONE 2019, 14, e0215204. [Google Scholar] [CrossRef]
- Taghizadeh, S.; Gholizadeh, M.; Rahimi-Mianji, G.; Moradi, M.H.; Costilla, R.; Moore, S.; Di Gerlando, R. Genome-wide identification of copy number variation and association with fat deposition in thin and fat-tailed sheep breeds. Sci. Rep. 2022, 12, 8834. [Google Scholar] [CrossRef]
- Ma, Q.; Liu, X.; Pan, J.; Ma, L.; Ma, Y.; He, X.; Zhao, Q.; Pu, Y.; Li, Y.; Jiang, L. Genome-wide detection of copy number variation in Chinese indigenous sheep using an ovine high-density 600 K SNP array. Sci. Rep. 2017, 7, 912. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, J.; Guo, Y.; Yang, Y.; Teng, T.; Yu, Q.; Wang, T.; Zhou, M.; Zhu, Q.; Wang, W. Genome-wide detection of CNVs and association with body weight in sheep based on 600K SNP arrays. Front. Genet. 2020, 11, 558. [Google Scholar] [CrossRef]
- Salehian-Dehkordi, H.; Xu, Y.-X.; Xu, S.-S.; Li, X.; Luo, L.-Y.; Liu, Y.-J.; Wang, D.-F.; Cao, Y.-H.; Shen, M.; Gao, L. Genome-wide detection of copy number variations and their association with distinct phenotypes in the world’s sheep. Front. Genet. 2021, 12, 670582. [Google Scholar] [CrossRef]
- Navarro Gonzalez, J.; Zweig, A.S.; Speir, M.L.; Schmelter, D.; Rosenbloom, K.R.; Raney, B.J.; Powell, C.C.; Nassar, L.R.; Maulding, N.D.; Lee, C.M. The UCSC genome browser database: 2021 update. Nucleic Acids Res. 2021, 49, D1046–D1057. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, L.; Xu, L.; Ren, H.; Lu, J.; Zhang, X.; Zhang, S.; Zhou, X.; Wei, C.; Zhao, F. Analysis of copy number variations in the sheep genome using 50K SNP BeadChip array. BMC Genom. 2013, 14, 229. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.M.; Goddard, M.E.; Black, M.A.; Brauning, R.; Auvray, B.; Dodds, K.G.; Kijas, J.W.; Cockett, N.; McEwan, J.C. Copy number variants in the sheep genome detected using multiple approaches. BMC Genom. 2016, 17, 441. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Cardone, M.F.; Wang, K.; Kim, E.-s.; Matukumalli, L.K.; Ventura, M.; Song, J.; VanRaden, P.M. Genomic characteristics of cattle copy number variations. BMC Genom. 2011, 12, 127. [Google Scholar] [CrossRef]
- Fontanesi, L.; Beretti, F.; Martelli, P.; Colombo, M.; Dall’Olio, S.; Occidente, M.; Portolano, B.; Casadio, R.; Matassino, D.; Russo, V. A first comparative map of copy number variations in the sheep genome. Genomics 2011, 97, 158–165. [Google Scholar] [CrossRef]
- Goyache, F.; Fernández, I.; Tapsoba, A.S.R.; Traoré, A.; Menéndez-Arias, N.A.; Álvarez, I. Functional characterization of Copy Number Variations regions in Djallonké sheep. J. Anim. Breed. Genet. 2021, 138, 600–612. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Q.; Lu, Z.; Zhao, X.; Zhang, Y. Analysis of copy number variations by SNP50 BeadChip array in Chinese sheep. Genomics 2015, 106, 295–300. [Google Scholar] [CrossRef]
- Chen, Y.; Bao, Z.; Liu, M.; Li, J.; Dai, Y.; Wang, F.; Zhang, X.; Zhai, P.; Zhao, B.; Wu, X. Promoter methylation changes in KRT17: A novel epigenetic marker for wool production in angora rabbit. Int. J. Mol. Sci. 2022, 23, 6077. [Google Scholar] [CrossRef]
- Cai, B.; Li, M.; Zheng, Y.; Yin, Y.; Jin, F.; Li, X.; Dong, J.; Jiao, X.; Liu, X.; Zhang, K. MicroRNA-149-Mediated MAPK1/ERK2 Suppression Attenuates Hair Follicle Stem Cell Differentiation. Hum. Gene Ther. 2022, 33, 625–637. [Google Scholar] [CrossRef]
- Liu, C.; He, Y.; Feng, X.; Li, J.; Wang, J. Expression of EPHA5 in lung adenocarcinoma is associated with lymph node metastasis and EGFR mutation. APMIS 2022, 130, 338–345. [Google Scholar] [CrossRef]
- Braga, L.G.; Chud, T.C.; Watanabe, R.N.; Savegnago, R.P.; Sena, T.M.; do Carmo, A.S.; Machado, M.A.; Panetto, J.C.d.C.; da Silva, M.V.G.; Munari, D.P. Identification of copy number variations in the genome of Dairy Gir cattle. PLoS ONE 2023, 18, e0284085. [Google Scholar] [CrossRef]
- Rees, E.; Kirov, G. Copy number variation and neuropsychiatric illness. Curr. Opin. Genet. Dev. 2021, 68, 57–63. [Google Scholar] [CrossRef]
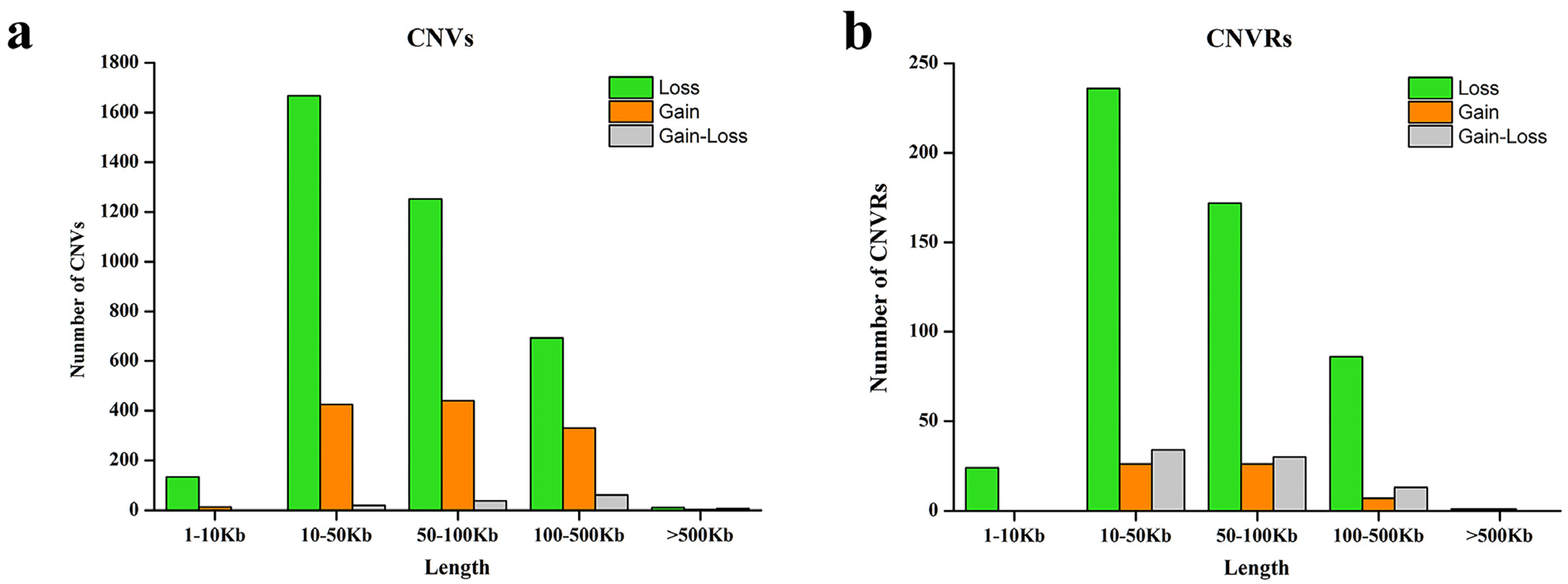

| Type | CNV | CNVR |
|---|---|---|
| Total number | 5101 | 656 |
| Average no. of CNVs per individual | 17.9 | 2.3 |
| CNV size per individual | 1.4 Mb | 0.2 Mb |
| Loss | 3760 | 519 |
| Gain | 1214 | 60 |
| Gain–loss | 127 | 77 |
| Total length | 388.1 Mbs | 43.9 Mbs |
| Average size | 76.1 Kbs | 66.9 Kbs |
| Median size | 55.2 Kbs | 51.1 Kbs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; An, J.; Zhang, X.; Di, J.; He, J.; Yasen, A.; Ma, Y.; Sailikehan, G.; Huang, X.; Tian, K. Genome-Wide Scan for Copy Number Variations in Chinese Merino Sheep Based on Ovine High-Density 600K SNP Arrays. Animals 2024, 14, 2897. https://doi.org/10.3390/ani14192897
Tian Y, An J, Zhang X, Di J, He J, Yasen A, Ma Y, Sailikehan G, Huang X, Tian K. Genome-Wide Scan for Copy Number Variations in Chinese Merino Sheep Based on Ovine High-Density 600K SNP Arrays. Animals. 2024; 14(19):2897. https://doi.org/10.3390/ani14192897
Chicago/Turabian StyleTian, Yuezhen, Jing An, Xinning Zhang, Jiang Di, Junmin He, Ayinuer Yasen, Yanpin Ma, Gaohaer Sailikehan, Xixia Huang, and Kechuan Tian. 2024. "Genome-Wide Scan for Copy Number Variations in Chinese Merino Sheep Based on Ovine High-Density 600K SNP Arrays" Animals 14, no. 19: 2897. https://doi.org/10.3390/ani14192897