
Genomic Insights and Conservation Priorities for Kongshan Cattle: A Whole-Genome Resequencing Approach
 , and
, and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction and Sequencing
2.2. SNP and InDel Calling
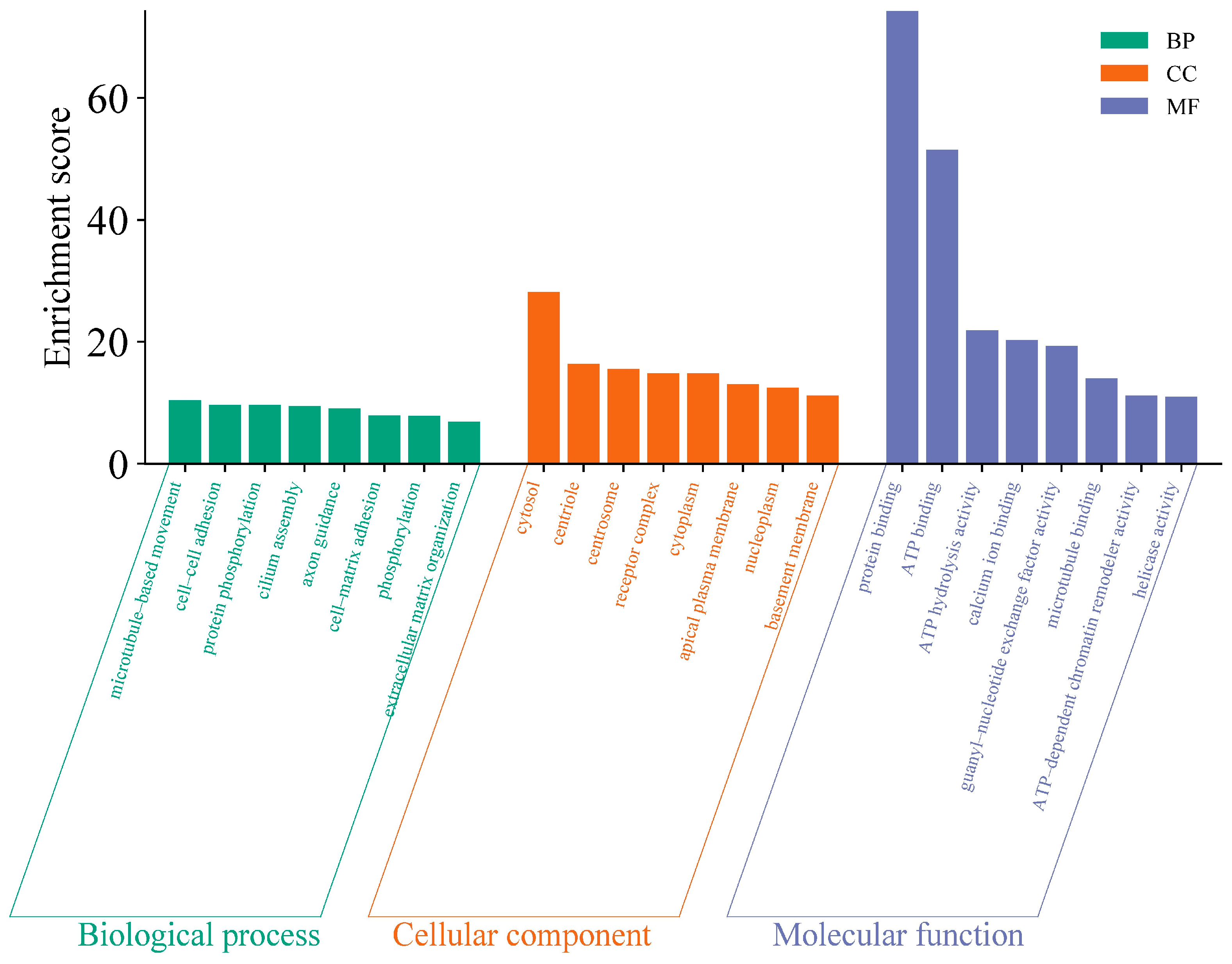
2.3. Functional Analysis of Variant Genes
2.4. Statistical Analyses
3. Results
3.1. Comprehensive Quality Assessment of Resequencing Data for Kongshan Cattle
3.2. Genomic SNP Variant Analysis: Transition, Transversion, and Zygosity Rates
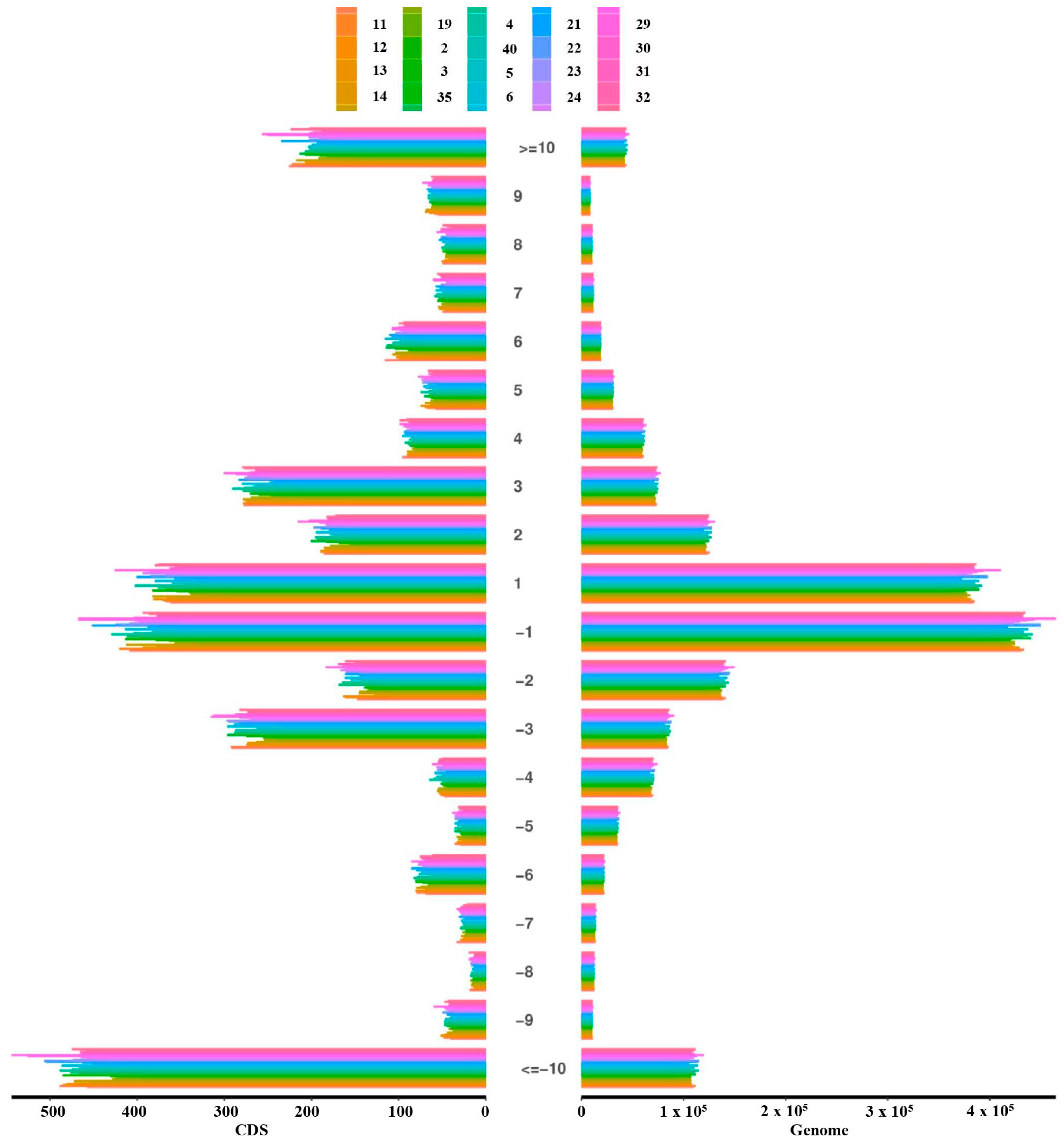
3.3. Analysis of Fragment Insertions and Deletions (InDels) in Genomic and Coding Regions
3.4. Mining Genetic Variations at the DNA Level
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, F.; Wang, W.; Shi, Y.; Fang, D.; Aguo, Y.; Gan, J.; Deng, X.; Fu, M.; Yi, J. Growth and Development Analysis of Kongshan Cattle. Agric. Biotechnol. 2022, 11, 62–65. [Google Scholar]
- Bayes, M.; Heath, S.; Gut, I.G. Applications of second generation sequencing technologies in complex disorders. Curr. Top. Behav. Neurosci. 2012, 12, 321–343. [Google Scholar] [PubMed]
- Martin, J.; Schackwitz, W.; Lipzen, A. Genomic Sequence Variation Analysis by Resequencing. Methods Mol. Biol. 2018, 1775, 229–239. [Google Scholar]
- Iqbal, N.; Liu, X.; Yang, T.; Huang, Z.; Hanif, Q.; Asif, M.; Khan, Q.M.; Mansoor, S. Genomic variants identified from whole-genome resequencing of indicine cattle breeds from Pakistan. PLoS ONE 2019, 14, e0215065. [Google Scholar] [CrossRef]
- Peripolli, E.; Reimer, C.; Ha, N.T.; Geibel, J.; Machado, M.A.; Panetto, J.; do Egito, A.A.; Baldi, F.; Simianer, H.; da Silva, M. Genome-wide detection of signatures of selection in indicine and Brazilian locally adapted taurine cattle breeds using whole-genome re-sequencing data. BMC Genom. 2020, 21, 624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yao, Z.; Li, X.; Zhang, Z.; Liu, X.; Yang, P.; Chen, N.; Xia, X.; Lyu, S.; Shi, Q.; et al. Assessing genomic diversity and signatures of selection in Pinan cattle using whole-genome sequencing data. BMC Genom. 2022, 23, 460. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, J.; Shen, M.; Xie, X.L.; Liu, G.J.; Xu, Y.X.; Lv, F.H.; Yang, H.; Yang, Y.L.; Liu, C.B.; et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 2815. [Google Scholar] [CrossRef]
- Li, X.; Ye, J.; Han, X.; Qiao, R.; Li, X.; Lv, G.; Wang, K. Whole-genome sequencing identifies potential candidate genes for reproductive traits in pigs. Genomics 2020, 112, 199–206. [Google Scholar] [CrossRef]
- Rubin, C.J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef]
- Mei, C.; Wang, H.; Liao, Q.; Wang, L.; Cheng, G.; Wang, H.; Zhao, C.; Zhao, S.; Song, J.; Guang, X.; et al. Genetic Architecture and Selection of Chinese Cattle Revealed by Whole Genome Resequencing. Mol. Biol. Evol. 2018, 35, 688–699. [Google Scholar] [CrossRef]
- Guan, X.; Zhao, S.; Xiang, W.; Jin, H.; Chen, N.; Lei, C.; Jia, Y.; Xu, L. Genetic Diversity and Selective Signature in Dabieshan Cattle Revealed by Whole-Genome Resequencing. Biology 2022, 11, 1327. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, S.; Zhang, H.; Zhang, Z.; Chen, N.; Li, Z.; Sun, H.; Liu, X.; Lyu, S.; Wang, X.; et al. Assessing genomic diversity and signatures of selection in Jiaxian Red cattle using whole-genome sequencing data. BMC Genom. 2021, 22, 43. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cheng, H.; Liu, Y.; Sun, L.; Chen, N.; Jiang, F.; You, W.; Yang, Z.; Zhang, B.; Song, E.; et al. Assessing Genomic Diversity and Selective Pressures in Bohai Black Cattle Using Whole-Genome Sequencing Data. Animals 2022, 12, 665. [Google Scholar] [CrossRef]
- Mariadassou, M.; Ramayo-Caldas, Y.; Charles, M.; Féménia, M.; Renand, G.; Rocha, D. Detection of selection signatures in Limousin cattle using whole-genome resequencing. Anim. Genet. 2020, 51, 815–819. [Google Scholar] [CrossRef]
- Jiang, J.; Gao, Y.; Hou, Y.; Li, W.; Zhang, S.; Zhang, Q.; Sun, D. Whole-genome resequencing of Holstein bulls for indel discovery and identification of genes associated with milk composition traits in dairy cattle. PLoS ONE 2016, 11, e0168946. [Google Scholar] [CrossRef]
- Berends, E.T.; Kuipers, A.; Ravesloot, M.M.; Urbanus, R.T.; Rooijakkers, S.H. Bacteria under stress by complement and coagulation. FEMS Microbiol. Rev. 2014, 38, 1146–1171. [Google Scholar] [CrossRef]
- Satyam, A.; Graef, E.R.; Lapchak, P.H.; Tsokos, M.G.; Dalle Lucca, J.J.; Tsokos, G.C. Complement and coagulation cascades in trauma. Acute Med. Surg. 2019, 6, 329–335. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Meira, L.B. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair 2006, 5, 189–209. [Google Scholar] [CrossRef]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Sample_ID | Clean_Reads | Clean_Base | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|
| 1 | 61,784,587 | 1.68 × 1010 | 98 | 94.74 | 43.08 |
| 2 | 59,958,158 | 1.595 × 1010 | 97.74 | 93.88 | 43.31 |
| 3 | 62,129,604 | 1.711 × 1010 | 98.01 | 94.82 | 42.91 |
| 4 | 65,090,871 | 1.812 × 1010 | 97.82 | 94.3 | 42.99 |
| 5 | 66,905,586 | 1.872 × 1010 | 97.87 | 94.38 | 42.53 |
| 6 | 60,576,373 | 1.618 × 1010 | 98.05 | 94.64 | 42.33 |
| 7 | 65,663,649 | 1.853 × 1010 | 97.67 | 93.89 | 42.36 |
| 8 | 65,313,818 | 1.816 × 1010 | 98.14 | 95.09 | 42.59 |
| 9 | 63,172,736 | 1.729 × 1010 | 98.18 | 95.17 | 43.14 |
| 10 | 67,870,038 | 1.926 × 1010 | 97.5 | 93.58 | 42.93 |
| 11 | 64,066,177 | 1.768 × 1010 | 97.91 | 94.79 | 43.17 |
| 12 | 63,900,471 | 1.764 × 1010 | 97.65 | 94.27 | 43.23 |
| 13 | 62,175,800 | 1.68 × 1010 | 97.71 | 93.78 | 42.58 |
| 14 | 57,245,601 | 1.5 × 1010 | 98.01 | 94.83 | 43.21 |
| 15 | 60,967,171 | 1.681 × 1010 | 97.82 | 94.4 | 43.11 |
| 16 | 64,245,777 | 1.754 × 1010 | 97.84 | 94.26 | 41.55 |
| 17 | 63,357,352 | 1.742 × 1010 | 98.12 | 94.93 | 42.7 |
| 18 | 64,428,527 | 1.791 × 1010 | 97.55 | 93.76 | 41.47 |
| 19 | 63,561,727 | 1.766 × 1010 | 97.76 | 94.11 | 42.17 |
| 20 | 54,372,159 | 1.553 × 1010 | 96.59 | 91.17 | 42.34 |
| 21 | 68,638,135 | 1.918 × 1010 | 98.11 | 95.15 | 43.45 |
| 22 | 66,325,088 | 1.851 × 1010 | 98.02 | 94.76 | 42.65 |
| 23 | 59,818,079 | 1.583 × 1010 | 97.94 | 94.59 | 43.61 |
| 24 | 66,007,821 | 1.841 × 1010 | 98.1 | 94.9 | 42.39 |
| 25 | 65,473,203 | 1.81 × 1010 | 98.23 | 95.32 | 42.82 |
| 26 | 64,494,351 | 1.746 × 1010 | 98.28 | 95.29 | 42.44 |
| 27 | 62,882,217 | 1.712 × 1010 | 97.96 | 94.62 | 42.93 |
| 28 | 67,728,437 | 1.887 × 1010 | 97.92 | 94.67 | 43.19 |
| 29 | 63,606,370 | 1.737 × 1010 | 97.87 | 94.17 | 42.75 |
| 29 | 63,606,370 | 1.737 × 1010 | 97.87 | 94.17 | 42.75 |
| 30 | 60,535,241 | 1.669 × 1010 | 97.41 | 93.11 | 42.45 |
| 31 | 63,851,243 | 1.742 × 1010 | 98.05 | 94.66 | 42.73 |
| 32 | 62,988,034 | 1.698 × 1010 | 98.13 | 94.98 | 41.81 |
| 33 | 64,991,465 | 1.811 × 1010 | 97.96 | 94.6 | 42.73 |
| 34 | 67,136,272 | 1.887 × 1010 | 97.78 | 94.15 | 42.83 |
| 35 | 65,230,266 | 1.813 × 1010 | 97.92 | 94.5 | 42.99 |
| 36 | 58,689,859 | 1.546 × 1010 | 98.06 | 94.82 | 43.41 |
| 37 | 60,724,316 | 1.638 × 1010 | 98.13 | 95.07 | 43.13 |
| 38 | 64,944,314 | 1.822 × 1010 | 97.85 | 94.26 | 42.53 |
| 39 | 68,160,352 | 1.93 × 1010 | 97.91 | 94.57 | 43.04 |
| 40 | 62,904,029 | 1.747 × 1010 | 97.72 | 94.08 | 40.95 |
| Mean | 63,547,882 | 1.75 × 1010 | 97.88 | 94.43 | 42.71 |
| Sample_ID | SNP Number | Transition | Transversion | Ti/Tv | Heterozygosity | Homozygosity | Hetratio |
|---|---|---|---|---|---|---|---|
| 1 | 14,207,746 | 10,064,021 | 4,143,725 | 2.42 | 3,383,344 | 10,824,402 | 23.81% |
| 2 | 13,240,910 | 9,379,967 | 3,860,943 | 2.42 | 2,774,942 | 10,465,968 | 20.95% |
| 3 | 13,917,295 | 9,861,512 | 4,055,783 | 2.43 | 3,089,436 | 10,827,859 | 22.19% |
| 4 | 14,023,735 | 9,933,509 | 4,090,226 | 2.42 | 3,187,450 | 10,836,285 | 22.72% |
| 5 | 14,218,607 | 10,070,821 | 4,147,786 | 2.42 | 3,418,603 | 10,800,004 | 24.04% |
| 6 | 13,284,189 | 9,399,724 | 3,884,465 | 2.41 | 2,774,123 | 10,510,066 | 20.88% |
| 7 | 14,454,668 | 10,243,981 | 4,210,687 | 2.43 | 3,313,682 | 11,140,986 | 22.92% |
| 8 | 13,665,815 | 9,670,487 | 3,995,328 | 2.42 | 2,993,846 | 10,671,969 | 21.90% |
| 9 | 13,796,846 | 9,777,019 | 4,019,827 | 2.43 | 3,157,604 | 10,639,242 | 22.88% |
| 10 | 14,330,750 | 10,154,645 | 4,176,105 | 2.43 | 3,541,413 | 10,789,337 | 24.71% |
| 11 | 14,096,969 | 9,984,328 | 4,112,641 | 2.42 | 3,306,860 | 10,790,109 | 23.45% |
| 12 | 14,029,291 | 9,938,041 | 4,091,250 | 2.42 | 3,173,186 | 10,856,105 | 22.61% |
| 13 | 14,046,698 | 9,950,937 | 4,095,761 | 2.42 | 3,354,423 | 10,692,275 | 23.88% |
| 14 | 13,623,689 | 9,657,310 | 3,966,379 | 2.43 | 3,238,142 | 10,385,547 | 23.76% |
| 15 | 13,893,159 | 9,841,825 | 4,051,334 | 2.42 | 2,561,481 | 11,331,678 | 18.43% |
| 16 | 13,861,477 | 9,800,903 | 4,060,574 | 2.41 | 2,625,420 | 11,236,057 | 18.94% |
| 17 | 13,752,486 | 9,734,535 | 4,017,951 | 2.42 | 2,886,794 | 10,865,692 | 20.99% |
| 18 | 13,893,767 | 9,823,070 | 4,070,697 | 2.41 | 3,327,115 | 10,566,652 | 23.94% |
| 19 | 13,958,597 | 9,887,685 | 4,070,912 | 2.42 | 3,370,129 | 10,588,468 | 24.14% |
| 20 | 13,006,597 | 9,216,408 | 3,790,189 | 2.43 | 2,850,516 | 10,156,081 | 21.91% |
| 21 | 14,669,911 | 10,395,409 | 4,274,502 | 2.43 | 3,701,319 | 10,968,592 | 25.23% |
| 22 | 14,672,998 | 10,394,392 | 4,278,606 | 2.42 | 3,753,107 | 10,919,891 | 25.57% |
| 23 | 13,703,719 | 9,716,393 | 3,987,326 | 2.43 | 3,042,421 | 10,661,298 | 22.20% |
| 24 | 13,868,570 | 9,815,622 | 4,052,948 | 2.42 | 3,195,874 | 10,672,696 | 23.04% |
| 25 | 14,100,902 | 9,991,380 | 4,109,522 | 2.43 | 3,155,358 | 10,945,544 | 22.37% |
| 26 | 14,254,834 | 10,092,102 | 4,162,732 | 2.42 | 3,558,844 | 10,695,990 | 24.96% |
| 27 | 14,576,978 | 10,331,589 | 4,245,389 | 2.43 | 3,705,680 | 10,871,298 | 25.42% |
| 28 | 15,117,210 | 10,711,301 | 4,405,909 | 2.43 | 3,892,154 | 11,225,056 | 25.74% |
| 29 | 14,278,695 | 10,119,132 | 4,159,563 | 2.43 | 3,468,084 | 10,810,611 | 24.28% |
| 30 | 13,888,549 | 9,843,688 | 4,044,861 | 2.43 | 3,244,361 | 10,644,188 | 23.35% |
| 31 | 14,177,372 | 10,040,463 | 4,136,909 | 2.42 | 3,435,967 | 10,741,405 | 24.23% |
| 32 | 14,129,984 | 9,995,601 | 4,134,383 | 2.41 | 3,403,540 | 10,726,444 | 24.08% |
| 33 | 14,251,061 | 10,098,826 | 4,152,235 | 2.43 | 3,486,580 | 10,764,481 | 24.46% |
| 34 | 14,305,678 | 10,138,099 | 4,167,579 | 2.43 | 3,456,719 | 10,848,959 | 24.16% |
| 35 | 14,433,117 | 10,220,758 | 4,212,359 | 2.42 | 3,579,334 | 10,853,783 | 24.79% |
| 36 | 13,417,037 | 9,508,151 | 3,908,886 | 2.43 | 2,252,453 | 11,164,584 | 16.78% |
| 37 | 14,195,922 | 10,055,439 | 4,140,483 | 2.42 | 3,222,503 | 10,973,419 | 22.70% |
| 38 | 14,373,091 | 10,183,304 | 4,189,787 | 2.43 | 3,692,607 | 10,680,484 | 25.69% |
| 39 | 14,571,879 | 10,327,152 | 4,244,727 | 2.43 | 3,555,457 | 11,016,422 | 24.39% |
| 40 | 14,044,682 | 9,920,111 | 4,124,571 | 2.4 | 3,529,280 | 10,515,402 | 25.12% |
| Mean | 14,058,387 | 9,957,241 | 4,101,146 | 2.42 | 3,266,504 | 10,791,883 | 23.19% |
| CDS | Genome | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Insertion | Deletion | Homo | Het | Total | Insertion | Deletion | Homo | Het | Total |
| 1 | 1467 | 1538 | 2317 | 688 | 3005 | 757,961 | 920,833 | 1,283,930 | 394,864 | 1,678,794 |
| 2 | 1358 | 1414 | 2165 | 607 | 2772 | 700,447 | 842,834 | 1,221,296 | 321,985 | 1,543,281 |
| 3 | 1459 | 1535 | 2315 | 679 | 2994 | 744,727 | 898,325 | 1,282,792 | 360,260 | 1,643,052 |
| 4 | 1423 | 1543 | 2267 | 699 | 2966 | 748,548 | 907,041 | 1,282,660 | 372,929 | 1,655,589 |
| 5 | 1408 | 1514 | 2207 | 715 | 2922 | 761,246 | 925,021 | 1,285,532 | 400,735 | 1,686,267 |
| 6 | 1190 | 1299 | 1972 | 517 | 2489 | 709,281 | 855,198 | 1,237,083 | 327,396 | 1,564,479 |
| 7 | 1497 | 1596 | 2363 | 730 | 3093 | 776,197 | 942,360 | 1,330,308 | 388,249 | 1,718,557 |
| 8 | 1292 | 1356 | 2072 | 576 | 2648 | 727,736 | 881,485 | 1,257,342 | 351,879 | 1,609,221 |
| 9 | 1394 | 1512 | 2210 | 696 | 2906 | 735,050 | 889,051 | 1,255,430 | 368,671 | 1,624,101 |
| 10 | 1426 | 1584 | 2278 | 732 | 3010 | 765,555 | 931,497 | 1,283,993 | 413,059 | 1,697,052 |
| 11 | 1474 | 1564 | 2350 | 688 | 3038 | 751,905 | 913,126 | 1,279,162 | 385,869 | 1,665,031 |
| 12 | 1485 | 1562 | 2354 | 693 | 3047 | 749,946 | 909,775 | 1,286,354 | 373,367 | 1,659,721 |
| 13 | 1352 | 1450 | 2122 | 680 | 2802 | 748,297 | 907,088 | 1,266,272 | 389,113 | 1,655,385 |
| 14 | 1438 | 1455 | 2279 | 614 | 2893 | 724,695 | 873,677 | 1,222,512 | 375,860 | 1,598,372 |
| 15 | 1484 | 1545 | 2447 | 582 | 3029 | 747,521 | 899,988 | 1,344,468 | 303,041 | 1,647,509 |
| 16 | 1186 | 1303 | 1975 | 514 | 2489 | 752,379 | 908,377 | 1,343,068 | 317,688 | 1,660,756 |
| 17 | 1382 | 1409 | 2156 | 635 | 2791 | 737,837 | 890,391 | 1,287,817 | 340,411 | 1,628,228 |
| 18 | 1188 | 1312 | 1907 | 593 | 2500 | 747,742 | 909,314 | 1,261,195 | 395,861 | 1,657,056 |
| 19 | 1295 | 1386 | 2059 | 622 | 2681 | 742,907 | 900,444 | 1,253,999 | 389,352 | 1,643,351 |
| 20 | 1268 | 1424 | 2057 | 635 | 2692 | 689,927 | 832,492 | 1,195,256 | 327,163 | 1,522,419 |
| 21 | 1550 | 1641 | 2449 | 742 | 3191 | 784,033 | 959,608 | 1,308,286 | 435,355 | 1,743,641 |
| 22 | 1479 | 1615 | 2304 | 790 | 3094 | 785,104 | 963,108 | 1,303,248 | 444,964 | 1,748,212 |
| 23 | 1433 | 1576 | 2278 | 731 | 3009 | 728,003 | 880,250 | 1,255,579 | 352,674 | 1,608,253 |
| 24 | 1295 | 1430 | 2114 | 611 | 2725 | 742,952 | 902,636 | 1,267,881 | 377,707 | 1,645,588 |
| 25 | 1463 | 1545 | 2305 | 703 | 3008 | 756,507 | 917,653 | 1,302,193 | 371,967 | 1,674,160 |
| 26 | 1382 | 1512 | 2187 | 707 | 2894 | 762,604 | 932,059 | 1,272,207 | 422,456 | 1,694,663 |
| 27 | 1565 | 1720 | 2481 | 804 | 3285 | 776,120 | 948,553 | 1,292,698 | 431,975 | 1,724,673 |
| 28 | 1658 | 1796 | 2568 | 886 | 3454 | 808,491 | 994,753 | 1,345,333 | 457,911 | 1,803,244 |
| 29 | 1456 | 1552 | 2240 | 768 | 3008 | 763,481 | 928,942 | 1,286,165 | 406,258 | 1,692,423 |
| 30 | 1387 | 1459 | 2192 | 654 | 2846 | 741,645 | 898,622 | 1,261,476 | 378,791 | 1,640,267 |
| 31 | 1385 | 1514 | 2209 | 690 | 2899 | 757,424 | 921,213 | 1,274,811 | 403,826 | 1,678,637 |
| 32 | 1255 | 1423 | 2001 | 677 | 2678 | 755,652 | 923,274 | 1,276,809 | 402,117 | 1,678,926 |
| 33 | 1473 | 1502 | 2287 | 688 | 2975 | 762,705 | 928,432 | 1,282,863 | 408,274 | 1,691,137 |
| 34 | 1444 | 1534 | 2276 | 702 | 2978 | 767,042 | 934,250 | 1,294,057 | 407,235 | 1,701,292 |
| 35 | 1473 | 1531 | 2282 | 722 | 3004 | 770,460 | 939,792 | 1,289,548 | 420,704 | 1,710,252 |
| 36 | 1398 | 1471 | 2298 | 571 | 2869 | 717,673 | 860,972 | 1,314,189 | 264,456 | 1,578,645 |
| 37 | 1482 | 1578 | 2337 | 723 | 3060 | 758,186 | 921,732 | 1,301,864 | 378,054 | 1,679,918 |
| 38 | 1451 | 1546 | 2289 | 708 | 2997 | 768,059 | 936,025 | 1,271,853 | 432,231 | 1,704,084 |
| 39 | 1518 | 1623 | 2338 | 803 | 3141 | 778,855 | 950,563 | 1,314,358 | 415,060 | 1,729,418 |
| 40 | 1218 | 1337 | 1926 | 629 | 2555 | 759,965 | 926,844 | 1,260,633 | 426,176 | 1,686,809 |
| Mean | 1405.775 | 1505.15 | 2230.825 | 680.1 | 2910.925 | 751,621.625 | 912,689.95 | 1,280,913 | 383,398.575 | 1,664,311.575 |
| Sample | Genes with Non-Synonymous SNP | Genes with InDel |
|---|---|---|
| 1 | 14,509 | 3350 |
| 10 | 14,553 | 3400 |
| 11 | 14,475 | 3386 |
| 12 | 14,556 | 3430 |
| 13 | 14,227 | 3188 |
| 14 | 14,312 | 3269 |
| 15 | 14,411 | 3397 |
| 16 | 13,665 | 2917 |
| 17 | 14,180 | 3191 |
| 18 | 13,506 | 2936 |
| 19 | 14,087 | 3102 |
| 2 | 14,077 | 3157 |
| 3 | 14,415 | 3371 |
| 35 | 14,525 | 3386 |
| 36 | 14,328 | 3199 |
| 37 | 14,603 | 3420 |
| 38 | 14,469 | 3335 |
| 39 | 14,598 | 3494 |
| 4 | 14,377 | 3287 |
| 40 | 13,453 | 2953 |
| 5 | 14,351 | 3270 |
| 6 | 13,619 | 2843 |
| 7 | 14,566 | 3459 |
| 8 | 13,963 | 3034 |
| 9 | 14,234 | 3244 |
| 20 | 13,826 | 3075 |
| 21 | 14,807 | 3530 |
| 22 | 14,638 | 3461 |
| 23 | 14,377 | 3335 |
| 24 | 14,045 | 3125 |
| 25 | 14,466 | 3398 |
| 26 | 14,214 | 3251 |
| 27 | 14,828 | 3603 |
| 28 | 15,089 | 3754 |
| 29 | 14,405 | 3345 |
| 30 | 14,254 | 3240 |
| 31 | 14,332 | 3251 |
| 32 | 13,817 | 3066 |
| 33 | 14,478 | 3358 |
| 34 | 14,460 | 3310 |
| Mean | 14,302.38 | 3278 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, W.; Ren, H.; Li, M.; Mei, L.; Zhang, B.; Jia, X.; Chen, S.; Wang, J.; Lai, S. Genomic Insights and Conservation Priorities for Kongshan Cattle: A Whole-Genome Resequencing Approach. Animals 2024, 14, 3056. https://doi.org/10.3390/ani14213056
Sun W, Ren H, Li M, Mei L, Zhang B, Jia X, Chen S, Wang J, Lai S. Genomic Insights and Conservation Priorities for Kongshan Cattle: A Whole-Genome Resequencing Approach. Animals. 2024; 14(21):3056. https://doi.org/10.3390/ani14213056
Chicago/Turabian StyleSun, Wenqiang, Hanjun Ren, Mengze Li, Liping Mei, Bingfei Zhang, Xianbo Jia, Shiyi Chen, Jie Wang, and Songjia Lai. 2024. "Genomic Insights and Conservation Priorities for Kongshan Cattle: A Whole-Genome Resequencing Approach" Animals 14, no. 21: 3056. https://doi.org/10.3390/ani14213056
APA StyleSun, W., Ren, H., Li, M., Mei, L., Zhang, B., Jia, X., Chen, S., Wang, J., & Lai, S. (2024). Genomic Insights and Conservation Priorities for Kongshan Cattle: A Whole-Genome Resequencing Approach. Animals, 14(21), 3056. https://doi.org/10.3390/ani14213056