
Effect of a Lactobacilli-Based Direct-Fed Microbial Product on Gut Microbiota and Gastrointestinal Morphological Changes

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Animals
2.2. Study Design and Animal Enrolment
2.3. Bacterial DNA Extraction from Faecal Samples
2.4. 16S rRNA Gene Amplicon Sequencing
2.5. Gross Pathology and Histopathology Examination
2.6. Alpha/Beta Diversity Analyses
2.7. Histopathology and Live Weight Data Analyses
3. Results
3.1. Live Weight Comparisons
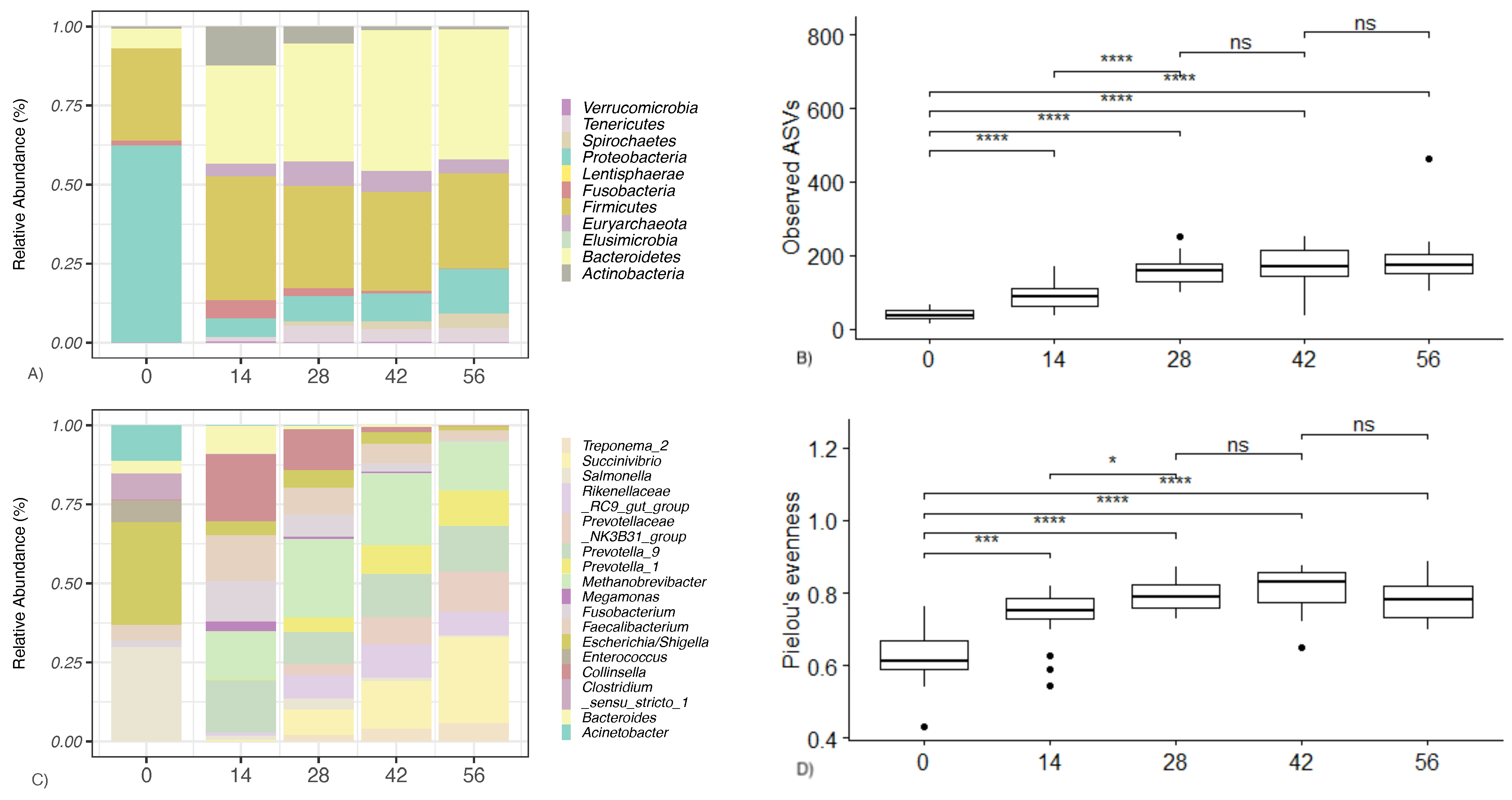
3.2. Sequencing Results
3.3. Compositional Differences between Different Groups
3.4. Metabolic Functions and Capacity of Ruminal and Faecal Microbiomes
3.5. Gross Pathology and Histopathology Examination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Du, W.; Wang, X.; Hu, M.; Hou, J.; Du, Y.; Si, W.; Yang, L.; Xu, L.; Xu, Q. Modulating gastrointestinal microbiota to alleviate diarrhea in calves. Front. Microbiol. 2023, 14, 1181545. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Elson, C.O. Adaptive immune education by gut microbiota antigens. Immunology 2018, 154, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Bian, B.; Teng, L.; Nelson, C.D.; Driver, J.; Elzo, M.A.; Jeong, K.C. Host genetic effects upon the early gut microbiota in a bovine model with graduated spectrum of genetic variation. ISME J. 2020, 14, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Torow, N.; Hornef, M.W. The neonatal window of opportunity: Setting the stage for life-long host-microbial interaction and immune homeostasis. J. Immunol. 2017, 198, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef]
- Amin, N.; Schwarzkopf, S.; Troscher-Mussotter, J.; Camarinha-Silva, A.; Danicke, S.; Huber, K.; Frahm, J.; Seifert, J. Host metabolome and faecal microbiome shows potential interactions impacted by age and weaning times in calves. Anim. Microbiome 2023, 5, 12. [Google Scholar] [CrossRef]
- Alipour, M.J.; Jalanka, J.; Pessa-Morikawa, T.; Kokkonen, T.; Satokari, R.; Hynonen, U.; Iivanainen, A.; Niku, M. The composition of the perinatal intestinal microbiota in cattle. Sci. Rep. 2018, 8, 10437–10451. [Google Scholar] [CrossRef]
- Dias, J.; Marcondes, M.I.; Motta de Souza, S.; Cardoso Da Mata E Silva, B.; Fontes Noronha, M.; Tassinari Resende, R.; Machado, F.S.; Cuquetto Mantovani, H.; Dill-Mcfarland, K.A.; Suen, G. Bacterial community dynamics across the gastrointestinal tracts of dairy calves during preweaning development. Appl. Environ. Microbiol. 2018, 84, 2675–2689. [Google Scholar] [CrossRef]
- Svetlana Ferreira, L.; de Sousa Bicalho, M.; Bicalho, R.C. The Bos taurus maternal microbiome: Role in determining the progeny early-life upper respiratory tract microbiome and health. PLoS ONE 2019, 14, e0208014. [Google Scholar] [CrossRef]
- Badman, J.; Daly, K.; Kelly, J.; Moran, A.W.; Cameron, J.; Watson, I.; Newbold, J.; Shirazi-Beechey, S.P. The effect of milk replacer composition on the intestinal microbiota of pre-ruminant dairy calves. Front. Vet. Sci. 2019, 6, 371. [Google Scholar] [CrossRef]
- Robertson, R.C.; Manges, A.R.; Finlay, B.B.; Prendergast, A.J. The Human microbiome and child growth-first 1000 days and beyond. Trends Microbiol. 2019, 27, 131–147. [Google Scholar] [CrossRef]
- Guilloteau, P.; Zabielski, R.; David, J.C.; Blum, J.W.; Morisset, J.A.; Biernat, M.; Wolinski, J.; Laubitz, D.; Hamon, Y. Sodium-butyrate as a growth promoter in milk replacer formula for young calves. J. Dairy Sci. 2009, 92, 1038–1049. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Griebel, P.J.; Guan, L.L. The gut microbiome and its potential role in the development and function of newborn calf gastrointestinal tract. Front. Vet. Sci. 2015, 2, 36. [Google Scholar] [CrossRef]
- Meale, S.J.; Li, S.C.; Azevedo, P.; Derakhshani, H.; DeVries, T.J.; Plaizier, J.C.; Steele, M.A.; Khafipour, E. Weaning age influences the severity of gastrointestinal microbiome shifts in dairy calves. Sci. Rep. 2017, 7, 198. [Google Scholar] [CrossRef] [PubMed]
- Bladwin VI, R.L.; McLeod, K.R.; Klotz, J.L.; Heitmann, R.N. Rumen development, intestinal growth and hepatic metabolism in the pre- and postweaning ruminant. J. Dairy Sci. 2004, 87, E55–E65. [Google Scholar] [CrossRef]
- Cezar, A.M.; Donde, S.C.; Tomaluski, C.R.; da Silva, A.P.; Toledo, A.F.; Coelho, M.G.; Virginio Junior, G.F.; Bittar, C.M.M. Age and post-prandial variations on selected metabolites in dairy calves fed different liquid diets. Animals 2022, 12, 3063. [Google Scholar] [CrossRef] [PubMed]
- Abecia, L.; Martin-Garcia, A.I.; Martinez, G.; Newbold, C.J.; Yanez-Ruiz, D.R. Nutritional intervention in early life to manipulate rumen microbial colonization and methane output by kid goats postweaning. J. Anim. Sci. 2013, 91, 4832–4840. [Google Scholar] [CrossRef] [PubMed]
- Meale, S.J.; Popova, M.; Saro, C.; Martin, C.; Bernard, A.; Lagree, M.; Yanez-Ruiz, D.R.; Boudra, H.; Duval, S.; Morgavi, D.P. Early life dietary intervention in dairy calves results in a long-term reduction in methane emissions. Sci. Rep. 2021, 11, 3003. [Google Scholar] [CrossRef]
- Jiao, J.; Wu, J.; Zhou, C.; Tang, S.; Wang, M.; Tan, Z. Composition of ileal bacterial community in grazing goats varies across non-rumination, transition and rumination stages of life. Front. Microbiol. 2016, 7, 1364. [Google Scholar] [CrossRef]
- Walter, J. Ecological role of lactobacilli in the gastrointestinal tract: Implications for fundamental and biomedical research. Appl. Environ. Microbiol. 2008, 74, 4985–4996. [Google Scholar] [CrossRef]
- Du, R.; Jiao, S.; Dai, Y.; An, J.; Lv, J.; Yan, X.; Wang, J.; Han, B. Probiotic Bacillus amyloliquefaciens C-1 improves growth performance, stimulates gh/igf-1, and regulates the gut microbiota of growth-retarded beef calves. Front. Microbiol. 2018, 9, 2006. [Google Scholar] [CrossRef]
- Kelly, A.; McCabe, M.; Kenny, D.; Waters, S. Effect of a butyrate-fortified milk replacer on gastrointestinal microbiota and products of fermentation in artificially reared dairy calves at weaning. Sci. Rep. 2018, 8, 14901. [Google Scholar] [CrossRef]
- Schofield, B.J.; Lachner, N.; Le, O.T.; McNeill, D.M.; Dart, P.; Ouwerkerk, D.; Hugenholtz, P.; Klieve, A.V. Beneficial changes in rumen bacterial community profile in sheep and dairy calves as a result of feeding the probiotic Bacillus amyloliquefaciens H57. J. Appl. Microbiol. 2018, 124, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Minato, H.; Otsuka, M.; Shirasaka, S.; Itabashi, H.; Mitsumori, M. Colonization of microorganisms in the rumen of young calves. J. Gen. Appl. Microbiol. 1992, 38, 447–456. [Google Scholar] [CrossRef]
- Barreto, M.O.; Soust, M.; Moore, R.J.; Olchowy, T.W.J.; Alawneh, J.I. Systematic review and meta-analysis of probiotic use on inflammatory biomarkers and disease prevention in cattle. Prev. Vet. Med. 2021, 194, 105433. [Google Scholar] [CrossRef] [PubMed]
- Virginio Junior, G.F.; Bittar, C.M.M. Microbial colonization of the gastrointestinal tract of dairy calves—A review of its importance and relationship to health and performance. Anim. Health Res. Rev. 2021, 22, 97–108. [Google Scholar] [CrossRef]
- Mansilla, F.I.; Ficoseco, C.A.; Miranda, M.H.; Puglisi, E.; Nader-Macias, M.E.F.; Vignolo, G.M.; Fontana, C.A. Administration of probiotic lactic acid bacteria to modulate fecal microbiome in feedlot cattle. Sci. Rep. 2022, 12, 12957. [Google Scholar] [CrossRef]
- Hewitt, A.; Olchowy, T.; James, A.S.; Fraser, B.; Ranjbar, S.; Soust, M.; Alawneh, J.I. Linear body measurements and productivity of subtropical Holstein-Friesian dairy calves. Aust. Vet. J. 2020, 98, 280–289. [Google Scholar] [CrossRef]
- Bielmann, V.; Gillan, J.; Perkins, N.R.; Skidmore, A.L.; Godden, S.; Leslie, K.E. An evaluation of Brix refractometry instruments for measurement of colostrum quality in dairy cattle. J. Dairy Sci. 2010, 93, 3713–3721. [Google Scholar] [CrossRef]
- Weaver, D.M.; Tyler, J.W.; VanMetre, D.C.; Hostetler, D.E.; Barrington, G.M. Passive transfer of colostral immunoglobulins in calves. J. Vet. Int. Med. 2000, 14, 569–577. [Google Scholar] [CrossRef]
- Chigerwe, M.; Tyler, J.W.; Schultz, L.G.; Middleton, J.R.; Steevens, B.J.; Spain, J.N. Effect of colostrum administration by use of oroesophageal intubation on serum IgG concentrations in Holstein bull calves. Am. J. Vet. Res. 2008, 69, 1158–1163. [Google Scholar] [CrossRef]
- Engelbrektson, A.; Kunin, V.; Wrighton, K.C.; Zvenigorodsky, N.; Chen, F.; Ochman, H.; Hugenholtz, P. Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J. 2010, 4, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.D.; Macklaim, J.M.; Linn, T.G.; Reid, G.; Gloor, G.B. ANOVA-Like differential gene expression analysis of single-organism and meta-RNA-Seq. PLoS ONE 2013, 8, e67019. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2012. [Google Scholar]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Du, J.; Yuan, Z.; Ma, Z.; Song, J.; Xie, X.; Chen, Y. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol. Biosyst. 2014, 10, 2441–2447. [Google Scholar] [CrossRef]
- Dohoo, I.R.; Martin, W.; Stryhn, W. Veterinary Epidemiologic Research; AVC Inc.: Charlottetown, QC, Canada, 2009. [Google Scholar]
- Stevenson, M.; Nunes, T.; Sanchez, J.; Thornton, R. epiR: Functions for Analysing Epidemiological Data. R Package, Version 0.9-3; 2008. Epi Center, Massey University, New Zealand. Available online: http://epicentre.massey.ac.nz (accessed on 1 April 2018).
- Bates, D. lme4: Linear Mixed-Effects Models Using S4 Classes. R Package, Version 0.99875-9. 2007. Available online: https://cran.r-project.org/web/packages/lme4/lme4.pdf (accessed on 15 February 2024).
- Kenez, A.; Koch, C.; Korst, M.; Kesser, J.; Eder, K.; Sauerwein, H.; Huber, K. Different milk feeding intensities during the first 4 weeks of rearing dairy calves: Part 3: Plasma metabolomics analysis reveals long-term metabolic imprinting in Holstein heifers. J. Dairy Sci. 2018, 101, 8446–8460. [Google Scholar] [CrossRef]
- Oikonomou, G.; Teixeira, A.G.V.; Foditsch, C.; Bicalho, M.L.; Machado, V.S.; Bicalho, R.C. Fecal microbial diversity in pre-weaned dairy calves as described by pyrosequencing of metagenomic 16S rDNA. Associations of Faecalibacterium species with health and growth. PLoS ONE 2013, 8, e63157. [Google Scholar] [CrossRef]
- Schwarzkopf, S.; Kinoshita, A.; Kluess, J.; Kersten, S.; Meyer, U.; Huber, K.; Dänicke, S.; Frahm, J. Weaning holstein calves at 17 weeks of age enables smooth transition from liquid to solid feed. Animals 2019, 9, 1132. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Griebel, P.J.; le Guan, L. Taxonomic identification of commensal bacteria associated with the mucosa and digesta throughout the gastrointestinal tracts of preweaned calves. Appl. Environ. Microbiol. 2014, 80, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.T.; Lee, S.J.; Kim, T.Y.; Lee, H.G.; Atikur, R.M.; Gu, B.H.; Kim, D.H.; Park, B.Y.; Son, J.K.; Kim, M.H. Dynamic changes in fecal microbial communities of neonatal dairy calves by aging and diarrhea. Animals 2021, 11, 1113. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, M.; Indugu, N.; Vecchiarelli, B.; Redding, L.; Bender, J.; Pappalardo, C.; Leibstein, M.; Toth, J.; Stefanovski, D.; Katepalli, A.; et al. Short communication: Comparison of the fecal bacterial communities in diarrheic and nondiarrheic dairy calves from multiple farms in southeastern Pennsylvania. J. Dairy Sci. 2021, 104, 7225–7232. [Google Scholar] [CrossRef] [PubMed]
- Maharshak, N.; Packey, C.D.; Ellermann, M.; Manick, S.; Siddle, J.P.; Huh, E.Y.; Plevy, S.; Sartor, R.B.; Carroll, I.M. Altered enteric microbiota ecology in interleukin 10-deficient mice during development and progression of intestinal inflammation. Gut Microbes 2013, 4, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Teal, T.K.; Marsh, T.L.; Tiedje, J.M.; Mosci, R.; Jernigan, K.; Zell, A.; Newton, D.W.; Salimnia, H.; Lephart, P.; et al. Intestinal microbial communities associated with acute enteric infections and disease recovery. Microbiome 2015, 3, 45. [Google Scholar] [CrossRef] [PubMed]
- Cappellozza, B.I.; Copani, G.; Boll, E.J.; Queiroz, O. Supplementation of direct-fed microbial Enterococcus faecium 669 affects performance of preweaning dairy calves. JDS Commun. 2023, 4, 284–287. [Google Scholar] [CrossRef]
- Roodposhti, P.M.; Dabiri, N. Effects of probiotic and prebiotic on average daily gain, fecal shedding of Escherichia Coli, and immune system status in newborn female calves. Asian-Australas. J. Anim. Sci. 2012, 25, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Sun, H.; Gao, H.; Xia, Y.; Zan, L.; Zhao, C. A meta-analysis on the effects of probiotics on the performance of pre-weaning dairy calves. J. Anim. Sci. Biotechnol. 2023, 14, 3. [Google Scholar] [CrossRef]
- Dick, K.J.; Duff, G.C.; Limesand, S.W.; Cuneo, S.P.; Knudson, D.K.; McMurphy, C.P.; Hall, L.W.; Bernal-Rigoli, J.C.; Marchello, J.A. Effects of a direct-fed microbial on digestive-tract morphology of Holstein bull calves and performance and carcass characteristics of Holstein steers. Prof. Anim. Sci. 2013, 29, 107–115. [Google Scholar] [CrossRef]
- Amin, N.; Seifert, J. Dynamic progression of the calf’s microbiome and its influence on host health. Comput. Struct. Biotechnol. J. 2021, 19, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Dill-McFarland, K.A.; Breaker, J.D.; Suen, G. Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci. Rep. 2017, 7, 40864. [Google Scholar] [CrossRef] [PubMed]
- Pilchova, T.; Pilet, M.F.; Cappelier, J.M.; Pazlarova, J.; Tresse, O. Protective effect of Carnobacterium spp. against Listeria monocytogenes during host cell invasion using in vitro HT29 model. Front. Cell Infect. Microbiol. 2016, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Józefiak, D.; Sip, A.; Kaczmarek, S.; Rutkowski, A. The effects of Carnobacterium divergens AS7 bacteriocin on gastrointestinal microflora in vitro and on nutrient retention in broiler chickens. J. Anim. Feed. Sci. 2010, 19, 460–467. [Google Scholar] [CrossRef]
- Pilet, M.F.; Dousset, X.; Barré, R.; Novel, G.; Desmazeaud, M.; Piard, J.C. Evidence for two bacteriocins produced by Carnobacterium pisciola and Carnobacterium divergens isolated from fish and active against Listeria monocytogenes. J. Food Prot. 1995, 58, 256–262. [Google Scholar] [CrossRef]
- Lesmeister, K.E.; Heinrichs, A.J. Effects of corn processing on growth characteristics, rumen development, and rumen parameters in neonatal dairy calves. J. Dairy Sci. 2004, 87, 3439–3450. [Google Scholar] [CrossRef]
- Yan, Z.; Zhang, K.; Zhang, K.; Wang, G.; Wang, L.; Zhang, J.; Qiu, Z.; Guo, Z.; Song, X.; Li, J. Integrated 16S rDNA Gene sequencing and untargeted metabolomics analyses to investigate the gut microbial composition and plasma metabolic phenotype in calves with dampness-heat diarrhea. Front. Vet. Sci. 2022, 9, 703051. [Google Scholar] [CrossRef]
- Wright, E.K.; Kamm, M.A.; Teo, S.M.; Inouye, M.; Wagner, J.; Kirkwood, C.D. Recent advances in characterizing the gastrointestinal microbiome in Crohn’s disease: A systematic review. Inflamm. Bowel Dis. 2015, 21, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Zhang, Y.; Fan, L.; Chen, J.; Wei, M.; Li, C.; Chen, X.; Zhang, L.; Yang, D.; Wang, J. Integrative analysis of vaginal microorganisms and serum metabolomics in rats with estrous cycle disorder induced by long-term heat exposure based on 16s rDNA gene sequencing and LC/MS-based metabolomics. Front. Cell Infect. Microbiol. 2021, 11, 595716. [Google Scholar] [CrossRef] [PubMed]
- Meale, S.J.; Li, S.; Azevedo, P.; Derakhshani, H.; Plaizier, J.C.; Khafipour, E.; Steele, M.A. Development of ruminal and fecal microbiomes are affected by weaning but not weaning strategy in dairy calves. Front. Microbiol. 2016, 7, 582. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alawneh, J.I.; Ramay, H.; Olchowy, T.; Allavena, R.; Soust, M.; Jassim, R.A. Effect of a Lactobacilli-Based Direct-Fed Microbial Product on Gut Microbiota and Gastrointestinal Morphological Changes. Animals 2024, 14, 693. https://doi.org/10.3390/ani14050693
Alawneh JI, Ramay H, Olchowy T, Allavena R, Soust M, Jassim RA. Effect of a Lactobacilli-Based Direct-Fed Microbial Product on Gut Microbiota and Gastrointestinal Morphological Changes. Animals. 2024; 14(5):693. https://doi.org/10.3390/ani14050693
Chicago/Turabian StyleAlawneh, John I., Hena Ramay, Timothy Olchowy, Rachel Allavena, Martin Soust, and Rafat Al Jassim. 2024. "Effect of a Lactobacilli-Based Direct-Fed Microbial Product on Gut Microbiota and Gastrointestinal Morphological Changes" Animals 14, no. 5: 693. https://doi.org/10.3390/ani14050693