
Comparative Genomics Reveal Phylogenetic Relationship and Chromosomal Evolutionary Events of Eight Cervidae Species

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Data
2.2. Phylogenetic Analysis

{kind=link}
{kind=link}
| Subfamily or Tribe | Species | Genome Size (Mb) | Scaffold N50 (Mb) | Haploid Karyotype |
|---|---|---|---|---|
| Cervini | Sika deer [24] (Cervus nippon) | 2500.64 | 78,786,809 | 32 + X |
| Red deer [25] (Cervus elaphus) | 2886.60 | 83,473,711 | 33 + X | |
| Tarim red deer (in press) (Cervus hanglu) | 2520.87 | 76.801.786 | 33 + X | |
| Wapiti [26] (Cervus canadensis) | 2526.61 | 77,654,944 | 33 + XY | |
| Capreolinae | Reindeer (Rangifer tarandus) | 2971.15 | 69,829,200 | 34 + XY |
| White-tailed deer [27] (Odocoileus virginianus) | 2380.49 | - | - | |
| Muntiacini | Reeves’ muntjac [28] (Muntiacus reevesi) | 2494.01 | 113,316,579 | 22 + X |
| Red muntjac [29] (Muntiacus muntjak) | 2489.50 | 682,452,208 | 2 + X | |
| Bovinae | Cattle [30] (Bos taurus) | 2770.67 | 103,308,737 | 29 + XY |
2.3. Divergence Time Estimation
2.4. Evolution of Gene Families
2.5. Chromosome Evolution
2.6. Gene Enrichment Analysis
3. Results and Discussions
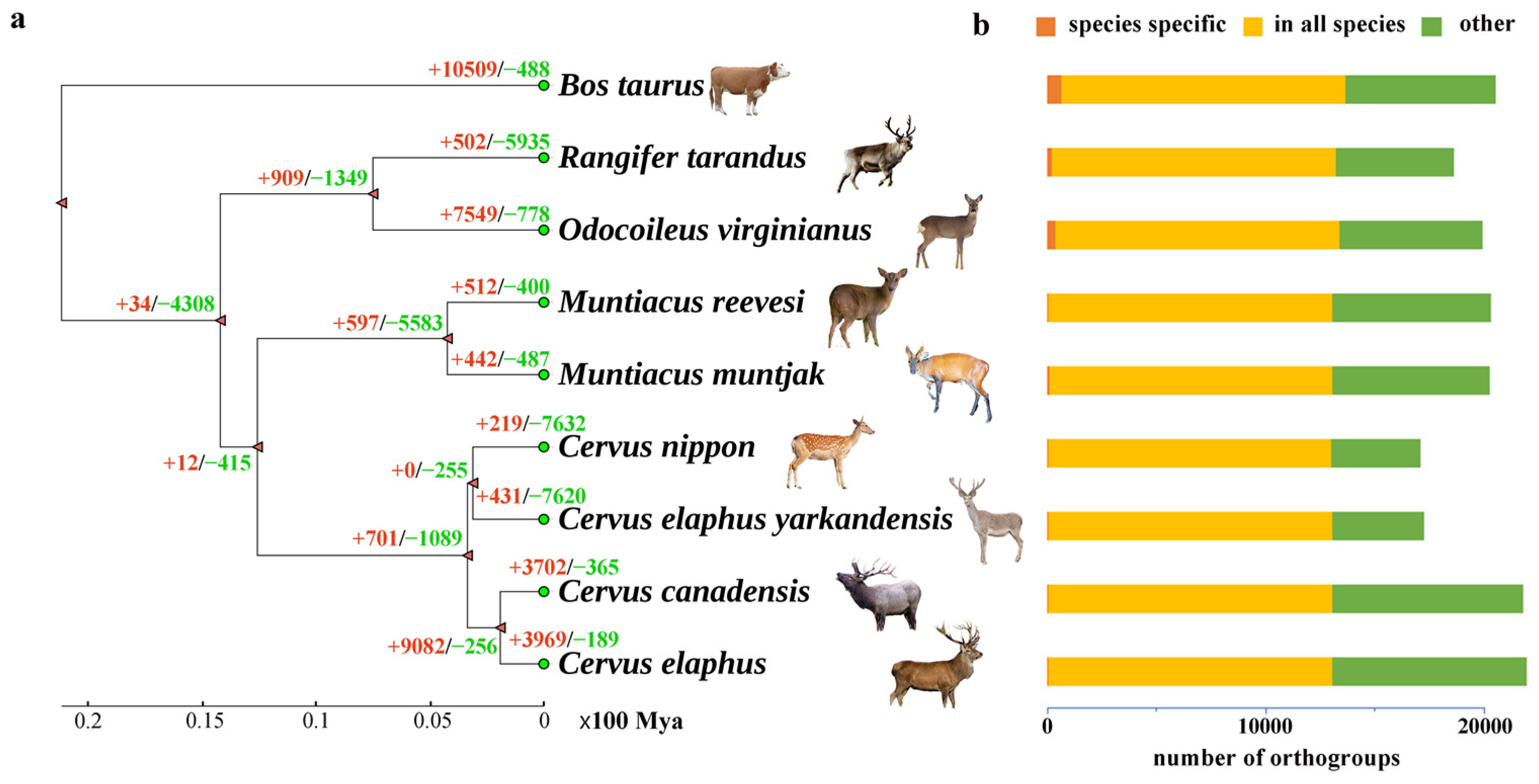
3.1. Phylogenetic Analysis
3.2. Divergence Time Estimating
3.3. Gene Family Evolution
3.4. Chromosome Evolution of Cervidae
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NCBI | National Center for Biotechnology Information |
| NGDC | National Genomics Data Center |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| CDS | Coding sequence |
| mtDNA | Mitochondrial DNA |
References
- Geist, V. Deer of the World: Their Evolution, Behavior, and Ecology. J. Wildl. Manag. 2000, 64, 606. [Google Scholar]
- Zhang, W.Q.; Zhang, M.H. Phylogeny and evolution of Cervidae based on complete mitochondrial genomes. Genet. Mol. Res. 2012, 11, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Ropiquet, A.; Hassanin, A. Mitochondrial and nuclear phylogenies of Cervidae (Mammalia, Ruminantia): Systematics, morphology, and biogeography. Mol. Phylogenet. Evol. 2006, 40, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Nishibori, M.; Yokohama, M. The complete nucleotide sequence of mitochondrial genome in the Japanese Sika deer (Cervus nippon), and a phylogenetic analysis between Cervidae and Bovidae. Small Rumin. Res. 2007, 69, 46–54. [Google Scholar] [CrossRef]
- Lovari, S.; Lorenzini, R.; Masseti, M.; Pereladova, O.; Carden, R.F.; Brook, S.M.; Mattioli, S. Cervus elaphus (errata version published in 2019). In The IUCN Red List of Threatened Species; International Union for Conservation of Nature: Gland, Switzerland, 2018; p. e.T55997072A142404453. [Google Scholar] [CrossRef]
- Brook, S.M.; Donnithorne-Tait, D.; Lorenzini, R.; Lovari, S.; Masseti, M.; Pereladova, O.; Ahmad, K.; Thakur, M. Cervus hanglu (amended version of 2017 assessment). In The IUCN Red List of Threatened Species; International Union for Conservation of Nature: Gland, Switzerland, 2017; p. e.T4261A120733024. [Google Scholar] [CrossRef]
- Brook, S.M.; Pluháček, J.; Lorenzini, R.; Lovari, S.; Masseti, M.; Pereladova, O.; Mattioli, S. Cervus canadensis (errata version published in 2019). In The IUCN Red List of Threatened Species; International Union for Conservation of Nature: Gland, Switzerland, 2018; p. e.T55997823A142396828. [Google Scholar] [CrossRef]
- Lorenzini, R.; Garofalo, L. Insights into the evolutionary history of Cervus (Cervidae, tribe Cervini) based on Bayesian analysis of mitochondrial marker sequences, with first indications for a new species. J. Zool. Syst. Evol. Res. 2015, 53, 340–349. [Google Scholar] [CrossRef]
- Mackiewicz, P.; Matosiuk, M.; Świsłocka, M.; Zachos, F.E.; Hajji, G.M.; Saveljev, A.P.; Seryodkin, I.V.; Farahvash, T.; Rezaei, H.R.; Torshizi, R.V.; et al. Phylogeny and evolution of the genus Cervus (Cervidae, Mammalia) as revealed by complete mitochondrial genomes. Sci. Rep. 2022, 12, 16381. [Google Scholar] [CrossRef] [PubMed]
- Kuwayama, R.; Ozawa, T. Phylogenetic relationships among european red deer, wapiti, and sika deer inferred from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2000, 15, 115–123. [Google Scholar] [CrossRef]
- Ludt, C.J.; Schroeder, W.; Rottmann, O.; Kuehn, R. Mitochondrial DNA phylogeography of red deer (Cervus elaphus). Mol. Phylogenet. Evol. 2004, 31, 1064–1083. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Rubini, M. Chromosomal evolution in Cervidae. Biosystems 1990, 24, 157–174. [Google Scholar] [CrossRef]
- Hu, X.; Jiang, Z.; Ming, Y.; Jian, J.; Jiang, S.; Zhang, D.; Zhang, J.; Zheng, S.; Fang, X.; Yang, Y. A chromosomal level genome sequence for Quasipaa spinosa (Dicroglossidae) reveals chromosomal evolution and population diversity. Mol. Ecol. Resour. 2021, 22, 1545–1558. [Google Scholar] [CrossRef]
- Hk, L.; Nsvall, K.; Vila, R.; Wiklund, C.; Backstrm, N. High-density linkage maps and chromosome level genome assemblies unveil direction and frequency of extensive structural rearrangements in wood white butterflies (Leptidea spp.). Chromosome Res. 2023, 31, 2. [Google Scholar] [CrossRef] [PubMed]
- Yamabe, T.; Kajitani, R.; Toyoda, A.; Itoh, T. Chromosomal-level Genome Assembly of the Coffee Bee Hawk Moth Reveals the Evolution of Chromosomes and the Molecular Basis of Distinct Phenotypes. Genome Biol. Evol. 2023, 15, evad141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, H.; Li, H.; Guo, J.; Ouyang, B.; Wang, M.; Xu, Q.; Wang, J.; Lv, M.; Guo, X.; et al. The White-Spotted Bamboo Shark Genome Reveals Chromosome Rearrangements and Fast-Evolving Immune Genes of Cartilaginous Fish. iScience 2020, 23, 101754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Ravi, V.; Qin, G.; Dai, H.; Zhang, H.X.; Han, F.M.; Wang, X.; Liu, Y.H.; Yin, J.P.; Huang, L.M.; et al. Comparative genomics reveal shared genomic changes in syngnathid fishes and signatures of genetic convergence with placental mammals. Natl. Sci. Rev. 2020, 7, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping. bioRxiv 2021. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Xing, X.; Ai, C.; Wang, T.; Li, Y.; Liu, H.; Hu, P.; Wang, G.; Liu, H.; Wang, H.; Zhang, R.; et al. The First High-quality Reference Genome of Sika Deer Provides Insights into High-tannin Adaptation. Genom. Proteom. Bioinform. 2023, 21, 203–215. [Google Scholar] [CrossRef]
- Bana, N.; Nyiri, A.; Nagy, J.; Frank, K.; Nagy, T.; Stéger, V.; Schiller, M.; Lakatos, P.; Sugár, L.; Horn, P.; et al. The red deer Cervus elaphus genome CerEla1.0: Sequencing, annotating, genes, and chromosomes. Mol. Genet. Genom. 2018, 293, 665–684. [Google Scholar] [CrossRef] [PubMed]
- Masonbrink, R.E.; Alt, D.; Bayles, D.O.; Boggiatto, P.; Edwards, W.; Tatum, F.; Williams, J.; Wilson-Welder, J.; Zimin, A.; Severin, A.; et al. A pseudomolecule assembly of the Rocky Mountain elk genome. PLoS ONE 2021, 16, e0249899. [Google Scholar] [CrossRef]
- London, E.W.; Roca, A.L.; Novakofski, J.E.; Mateus-Pinilla, N.E. A De Novo Chromosome-Level Genome Assembly of the White-Tailed Deer, Odocoileus virginianus. J. Hered. 2022, 113, 479–489. [Google Scholar] [CrossRef]
- Yin, Y.; Fan, H.; Zhou, B.; Hu, Y.; Fan, G.; Wang, J.; Zhou, F.; Nie, W.; Zhang, C.; Liu, L.; et al. Molecular mechanisms and topological consequences of drastic chromosomal rearrangements of muntjac deer. Nat. Commun. 2021, 12, 6858. [Google Scholar] [CrossRef]
- Mudd, A.B.; Bredeson, J.V.; Baum, R.; Hockemeyer, D.; Rokhsar, D.S. Analysis of muntjac deer genome and chromatin architecture reveals rapid karyotype evolution. Commun. Biol. 2020, 3, 480. [Google Scholar] [CrossRef]
- Vanvanhossou, S.F.U.; Yin, T.; Scheper, C.; Fries, R.; Dossa, L.H.; König, S. Unraveling Admixture, Inbreeding, and Recent Selection Signatures in West African Indigenous Cattle Populations in Benin. Front. Genet. 2021, 12, 657282. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2021, 36, 5516–5518. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Yang, J.; Jing, Y.; Xu, L.; Yu, K.; Fang, X. NGenomeSyn: An easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics 2023, 39, btad121. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Pitra, C.; Fickel, J.; Meijaard, E.; Groves, P.C. Evolution and phylogeny of old world deer. Mol. Phylogenet. Evol. 2004, 33, 880–895. [Google Scholar] [CrossRef] [PubMed]
- Heckeberg, N.S. The systematics of the Cervidae: A total evidence approach. PeerJ 2020, 8, e8114. [Google Scholar] [CrossRef] [PubMed]
- Randi, E.; Mucci, N.; Claro-Hergueta, F.; Bonnet, A.; Douzery, E.J.P. A mitochondrial DNA control region phylogeny of the Cervinae: Speciation in Cervus and implications for conservation. Anim. Conserv. 2010, 4, 1–11. [Google Scholar] [CrossRef]
- Chong-Zhen, Z.; Qing-Tao, Z.; Yuan-Huai, Q.; Yong, Y. Phylogenetic relationships among several genera in Drosophilidae deduced from mitochondrial cytochrome b gene partial sequence. J. Hubei Univ. (Nat. Sci. Ed.) 2004, 26, 344–349. [Google Scholar]
- Yuan, Y.; Zhang, Y.; Zhang, P.; Liu, C.; Wang, J.; Gao, H.; Hoelzel, A.R.; Seim, I.; Lv, M.; Lin, M.; et al. Comparative genomics provides insights into the aquatic adaptations of mammals. Proc. Natl. Acad. Sci. USA 2021, 118, e2106080118. [Google Scholar] [CrossRef]
- Chen, L.; Qiu, Q.; Jiang, Y.; Wang, K.; Lin, Z.; Li, Z.; Bibi, F.; Yang, Y.; Wang, J.; Nie, W.; et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 2019, 364, eaav6202. [Google Scholar] [CrossRef] [PubMed]
- Mahmut, H.; Masuda, R.; Onuma, M.; Takahashi, M.; Nagata, J.; Suzuki, M.; Ohtaishi, N. Molecular phylogeography of the red deer (Cervus elaphus) populations in Xinjiang of China: Comparison with other Asian, European, and North American populations. Zool. Sci. 2002, 19, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ba, H.; Yang, F. Complete mitochondrial genome of Cervus elaphus songaricus (Cetartiodactyla: Cervinae) and a phylogenetic analysis with related species. Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2016, 27, 620–621. [Google Scholar] [CrossRef]
- Perrière, G. Bioinformatics in the complete genome sequence era. Biochimie 2008, 90, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, P.K.; Pozio, E.; La Rosa, G.; Chang, B.C.H.; Koehler, A.V.; Hoberg, E.P.; Boag, P.R.; Tan, P.; Jex, A.R.; Hofmann, A.; et al. Phylogenomic and biogeographic reconstruction of the Trichinella complex. Nat. Commun. 2016, 7, 10513. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhou, J.; Zhou, J.; Hu, M.; Zhang, Q.; Kong, N.; Ren, H.; Liang, L.; Yue, J. Genome-based classification of Burkholderia cepacia complex provides new insight into its taxonomic status. Biol. Direct 2020, 15, 6. [Google Scholar] [CrossRef] [PubMed]
- Kapli, P.; Yang, Z.; Telford, M.J. Phylogenetic tree building in the genomic age. Nat. Rev. Genet. 2020, 21, 428–444. [Google Scholar] [CrossRef]
- O’Reilly, J.E.; Dos Reis, M.; Donoghue, P.C.J. Dating Tips for Divergence-Time Estimation. Trends Genet. 2015, 31, 637–650. [Google Scholar] [CrossRef]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef]
- Heath, T.A.; Huelsenbeck, J.P.; Stadler, T. The fossilized birth-death process for coherent calibration of divergence-time estimates. Proc. Natl. Acad. Sci. USA 2014, 111, E2957–E2966. [Google Scholar] [CrossRef]
- Xuan, J.L.; Scheffer, S.J.; Lewis, M.; Cassel, B.K.; Liu, W.X.; Wiegmann, B.M. The phylogeny and divergence times of leaf-mining flies (Diptera: Agromyzidae) from anchored phylogenomics. Mol. Phylogenet. Evol. 2023, 184, 107778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, S.; Xu, J.; Chen, B.; Zhou, K.; Yang, G. Phylogenomic analysis resolves the interordinal relationships and rapid diversification of the laurasiatherian mammals. Syst. Biol. 2012, 61, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Carretero, S.; Tamuri, A.U.; Battini, M.; Nascimento, F.F.; Carlisle, E.; Asher, R.J.; Yang, Z.; Donoghue, P.C.J.; Dos Reis, M. A species-level timeline of mammal evolution integrating phylogenomic data. Nature 2022, 602, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef] [PubMed]
- Mahony, S.; Foley, N.M.; Biju, S.D.; Teeling, E.C. Evolutionary History of the Asian Horned Frogs (Megophryinae): Integrative Approaches to Timetree Dating in the Absence of a Fossil Record. Mol. Biol. Evol. 2017, 34, 744–771. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.M. Gene factories, microfunctionalization and the evolution of gene families. Trends Genet. 2005, 21, 591–595. [Google Scholar] [CrossRef]
- Freitas, L.; Nery, M.F. Expansions and contractions in gene families of independently-evolved blood-feeding insects. BMC Evol. Biol. 2020, 20, 87. [Google Scholar] [CrossRef]
- Keller, M.; Baum, M.J.; Brock, O.; Brennan, P.A.; Bakker, J. The main and the accessory olfactory systems interact in the control of mate recognition and sexual behavior. Behav. Brain Res. 2009, 200, 268–276. [Google Scholar] [CrossRef]
- Wisenden, B.D.; Andebrhan, A.A.; Anderson, C.M.; Angus, J.M.; Coffman, I.C.; Cloutier, M.E.; Dahl, Q.H.; Doebler, A.D.; Erickson, L.G.; McKay, J.E.; et al. Olfactory cues of risk and visual cues of safety interact with sympatry and phylogeny in shaping behavioral responses by littoral fishes. Behav. Ecol. Sociobiol. 2023, 77, 91. [Google Scholar] [CrossRef]
- Hughes, G.M.; Boston, E.S.M.; Finarelli, J.A.; Murphy, W.J.; Higgins, D.G.; Teeling, E.C. The Birth and Death of Olfactory Receptor Gene Families in Mammalian Niche Adaptation. Mol. Biol. Evol. 2018, 35, 1390–1406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Ma, J.Z. A Theoretical Review of Wildlife Habitat Fragmentation. Chin. J. Wildl. 2014, 35, 6–14. [Google Scholar] [CrossRef]
- Haddad, N.M.; Brudvig, L.A.; Clobert, J.; Davies, K.F.; Gonzalez, A.; Holt, R.D.; Lovejoy, T.E.; Sexton, J.O.; Austin, M.P.; Collins, C.D.; et al. Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci. Adv. 2015, 1, e1500052. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wen, X.; Zhang, R.; Xing, X. Current Situation and Utilization of Velvet Deer Germplasm Resources in China. Animals 2022, 12, 3529. [Google Scholar] [CrossRef]
- Zhang, B. Wildlife of Cervidae in China. Biol. Teach. 2008, 6, 4–6. [Google Scholar]
- Ba, H.; Qin, T.; Cai, Z.; Liu, W.; Li, C. Molecular evidence for adaptive evolution of olfactory-related genes in cervids. Genes Genom. 2020, 42, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Bártová, E.; Krejcí, J.; Harnicarová, A.; Galiová, G.; Kozubek, S. Histone modifications and nuclear architecture: A review. J. Histochem. Cytochem. 2008, 56, 711–721. [Google Scholar] [CrossRef]
- Homberg, M.; Magin, T.M. Beyond expectations: Novel insights into epidermal keratin function and regulation. Int. Rev. Cell Mol. Biol. 2014, 311, 265–306. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.D.; Irwin, D.M.; Zhang, Y.P. Molecular evolution of the keratin associated protein gene family in mammals, role in the evolution of mammalian hair. BMC Evol. Biol. 2008, 8, 241. [Google Scholar] [CrossRef]
- Maderson, P.F. Mammalian skin evolution: A reevaluation. Exp. Dermatol. 2003, 12, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, F.; Fischer, H.; Langbein, L.; Praetzel-Wunder, S.; Ebner, B.; Figlak, K.; Weissenbacher, A.; Sipos, W.; Tschachler, E.; Eckhart, L. Differential Evolution of the Epidermal Keratin Cytoskeleton in Terrestrial and Aquatic Mammals. Mol. Biol. Evol. 2019, 36, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, J.; Kubickova, S.; Musilova, P.; Cernohorska, H.; Muskova, H.; Vodicka, R.; Rubes, J. Karyotype relationships among selected deer species and cattle revealed by bovine FISH probes. PLoS ONE 2017, 12, e0187559. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Han, L.; Zhao, X.; Wang, Q.; Xia, Y.; Li, H. Haplotype-resolved Genome of Sika Deer Reveals Allele-specific Gene Expression and Chromosome Evolution. Genom. Proteom. Bioinform. 2023, 21, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Du, D.R. Karyotypes of Cervidae and their evolution. Acta Zool. Sin. 1983, 29, 214–222. [Google Scholar]
- Yang, F.; O’Brien, P.C.M.; Wienberg, J.; Ferguson-Smith, M.A. A reappraisal of the tandem fusion theory of karyotype evolution in the Indian muntjac using chromosome painting. Chromosome Res. 1997, 5, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.F.; Shan, X.N.; Li, J.; Shi, T.Y.; Zheng, A.L. Sequence analysis and phylogeny of deer (Cervidae) MtDNA control regions. Acta Genet. Sin. 2004, 31, 395–402. [Google Scholar] [PubMed]
- Frönicke, L.; Chowdhary, B.P.; Scherthan, H. Segmental homology among cattle (Bos taurus), Indian muntjac (Muntiacus muntjak vaginalis), and Chinese muntjac (M. reevesi) karyotypes. Cytogenet. Cell Genet. 1997, 77, 223–227. [Google Scholar] [CrossRef]
- Huang, L.; Wang, J.; Nie, W.; Su, W.; Yang, F. Tandem chromosome fusions in karyotypic evolution of Muntiacus: Evidence from M. feae and M. gongshanensis. Chromosome Res. 2006, 14, 637–647. [Google Scholar] [CrossRef]
- Yang, F.; Carter, N.P.; Shi, L.; Ferguson-Smith, M.A. A comparative study of karyotypes of muntjacs by chromosome painting. Chromosoma 1995, 103, 642–652. [Google Scholar] [CrossRef]

| Species | Contractive | Expansive | ||
|---|---|---|---|---|
| Gene Families | Genes | Gene Families | Genes | |
| Reindeer | 1795 | 1917 | 108 | 1434 |
| White-tailed deer | 162 | 273 | 1690 | 12,418 |
| Reeves’ muntjac | 136 | 365 | 126 | 1285 |
| Red muntjac | 138 | 315 | 126 | 1230 |
| Red deer | 32 | 70 | 2109 | 19,456 |
| Wapiti | 67 | 144 | 1994 | 18,075 |
| Tarim red deer | 2212 | 2121 | 25 | 202 |
| Sika deer | 2202 | 2127 | 21 | 286 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, L.; Dong, S.; Xing, X. Comparative Genomics Reveal Phylogenetic Relationship and Chromosomal Evolutionary Events of Eight Cervidae Species. Animals 2024, 14, 1063. https://doi.org/10.3390/ani14071063
Tang L, Dong S, Xing X. Comparative Genomics Reveal Phylogenetic Relationship and Chromosomal Evolutionary Events of Eight Cervidae Species. Animals. 2024; 14(7):1063. https://doi.org/10.3390/ani14071063
Chicago/Turabian StyleTang, Lixin, Shiwu Dong, and Xiumei Xing. 2024. "Comparative Genomics Reveal Phylogenetic Relationship and Chromosomal Evolutionary Events of Eight Cervidae Species" Animals 14, no. 7: 1063. https://doi.org/10.3390/ani14071063