
Genomic Prediction of Growth Traits in Yorkshire Pigs of Different Reference Group Sizes Using Different Estimated Breeding Value Models
 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Husbandry Management of Experimental Animals
2.2. Trait-Corrected Models for Experimental Animals
2.3. DNA Extraction and Genotyping
2.4. Genomic Analysis of the Population
2.5. Scenarios of Combining References
2.6. Statistical Analysis
2.6.1. Bayesian A and Bayesian B
2.6.2. BayesC
2.6.3. GBLUP
2.6.4. ssGBLUP
2.7. Evaluation of the Accuracy of Genomic Prediction
3. Results
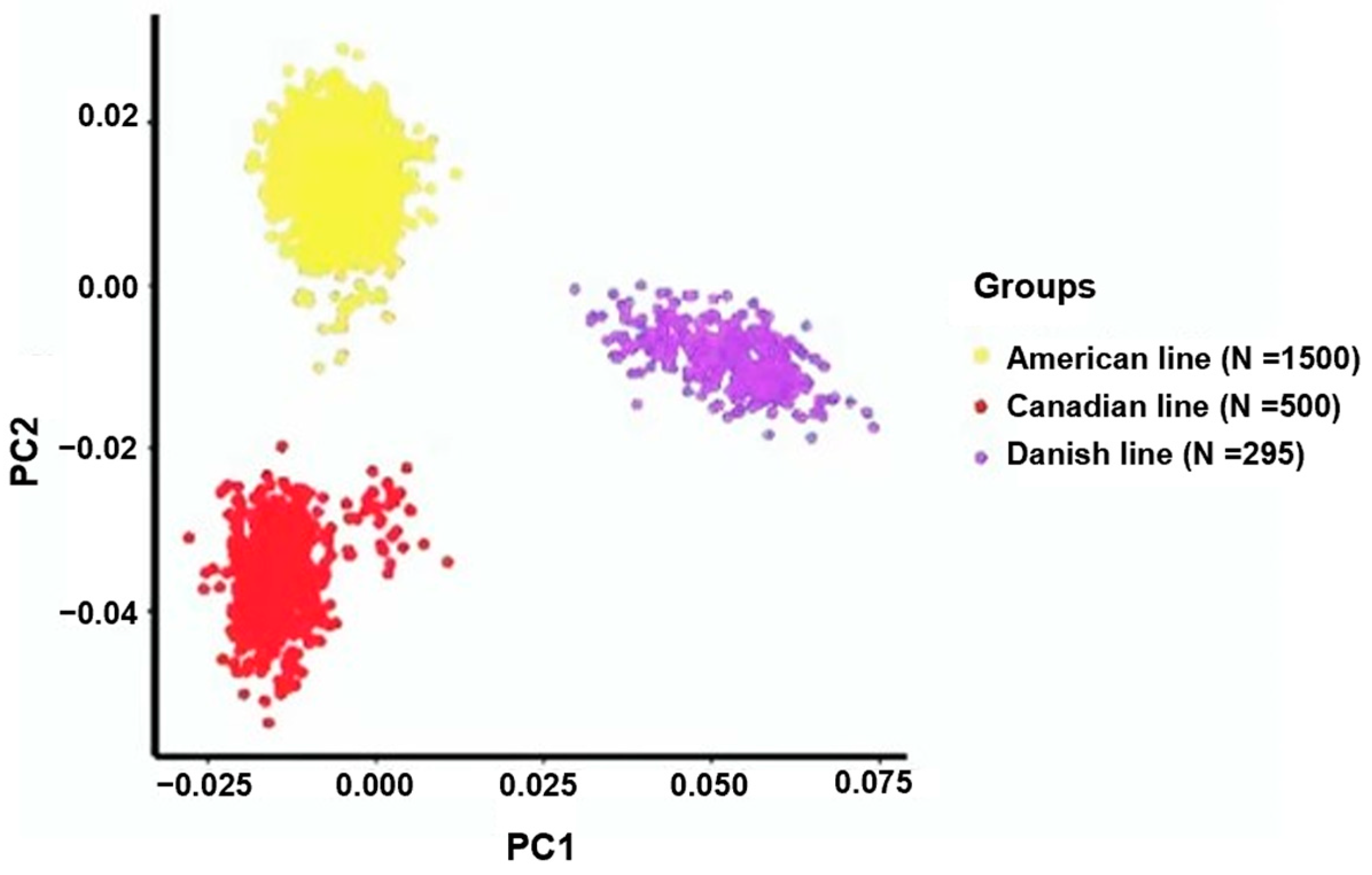
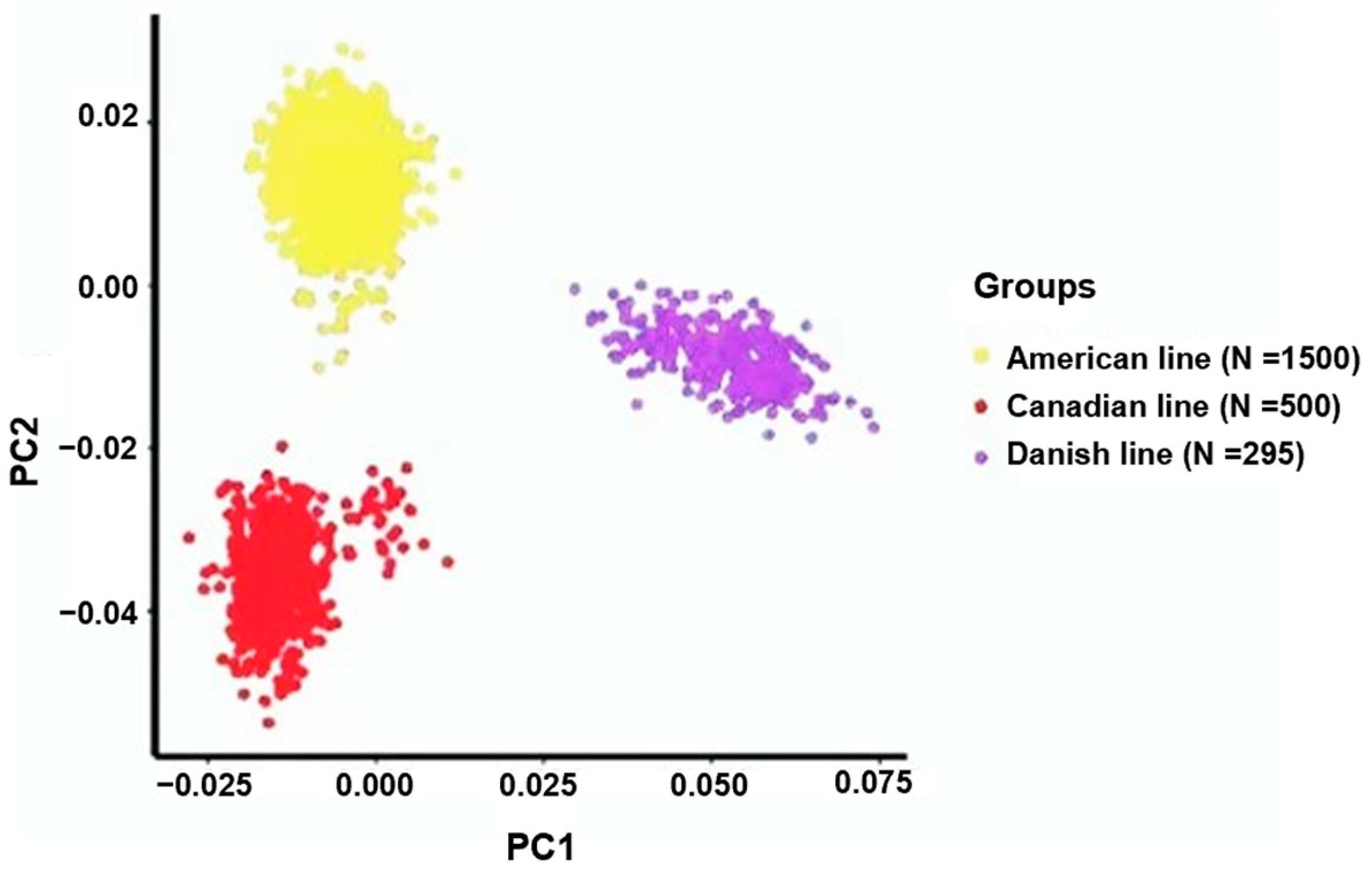
3.1. Assessment of Population Stratification
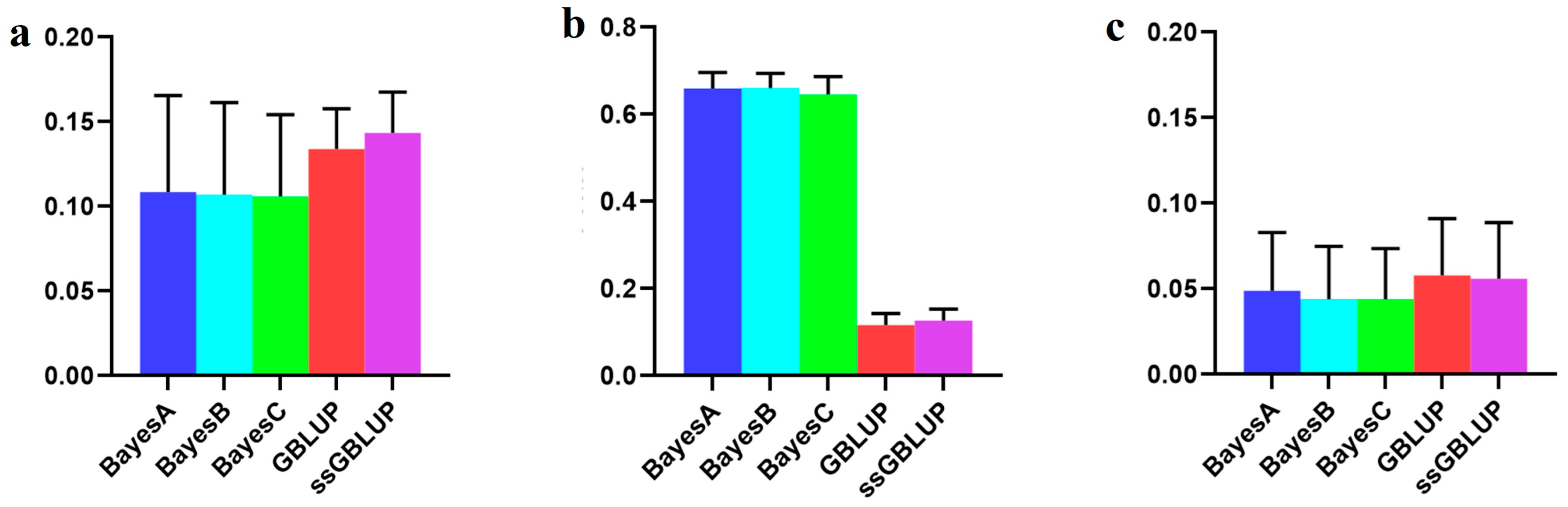
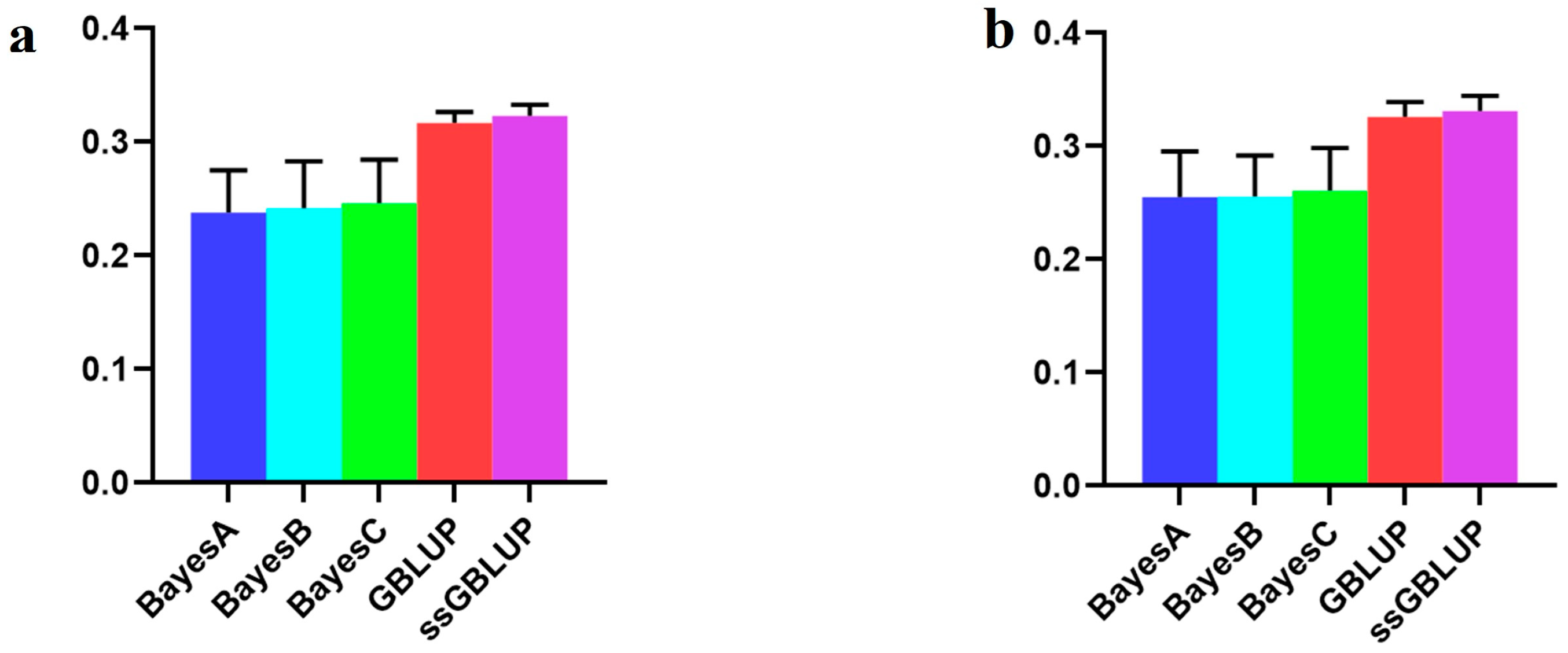
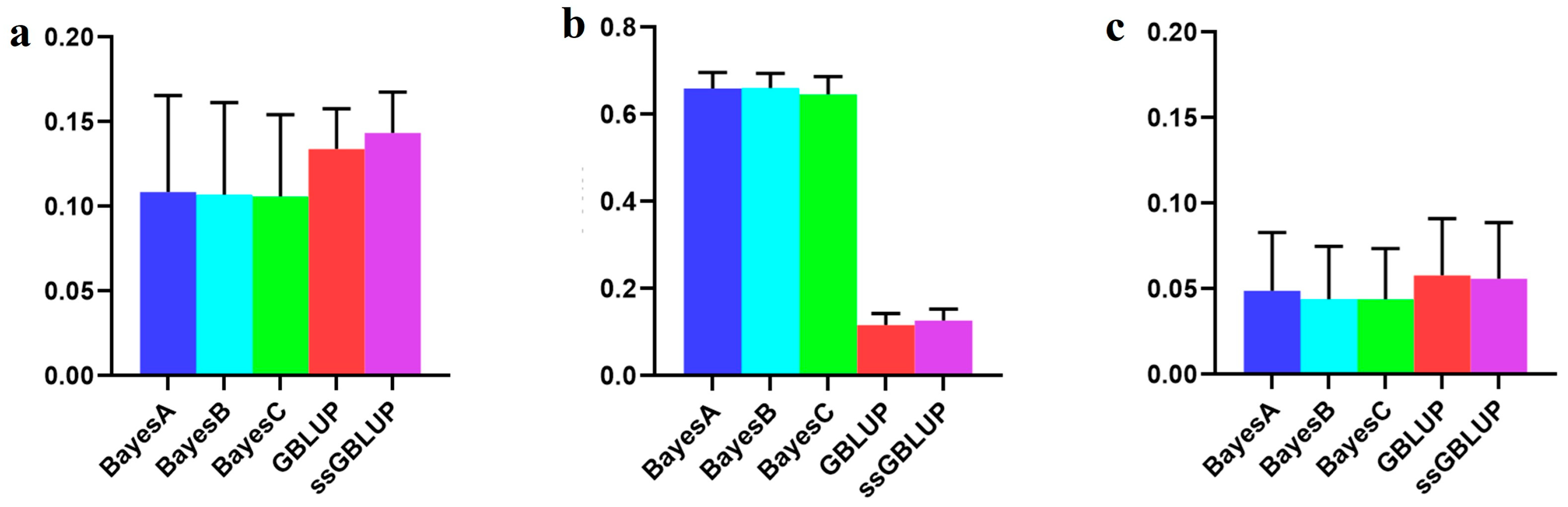
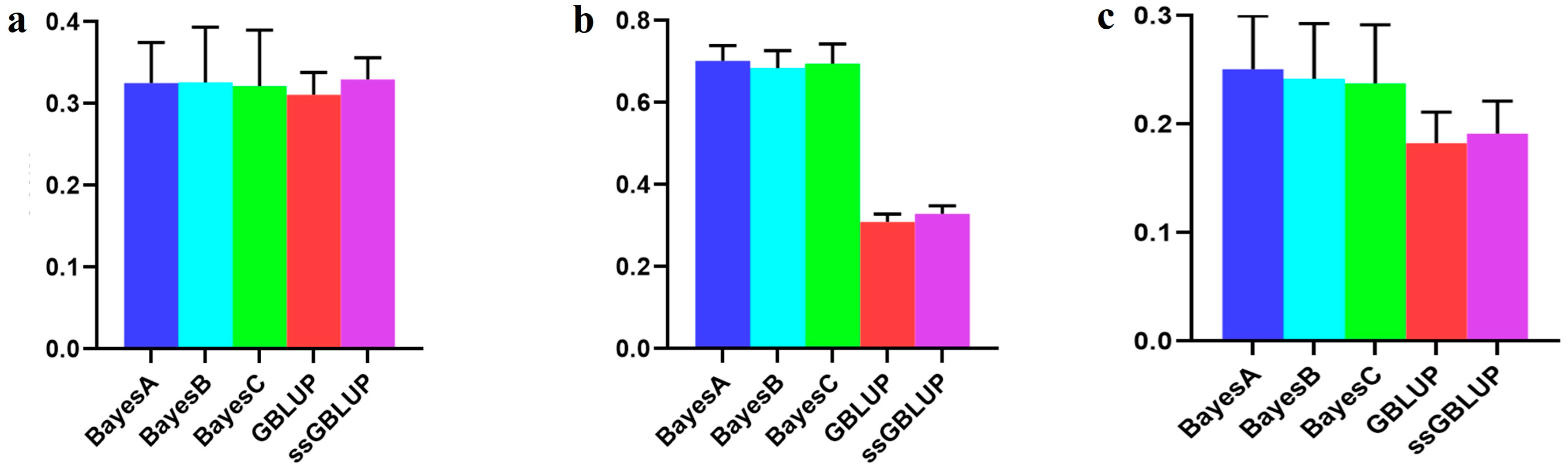
3.2. The Accuracy of the Danish (Small) Line
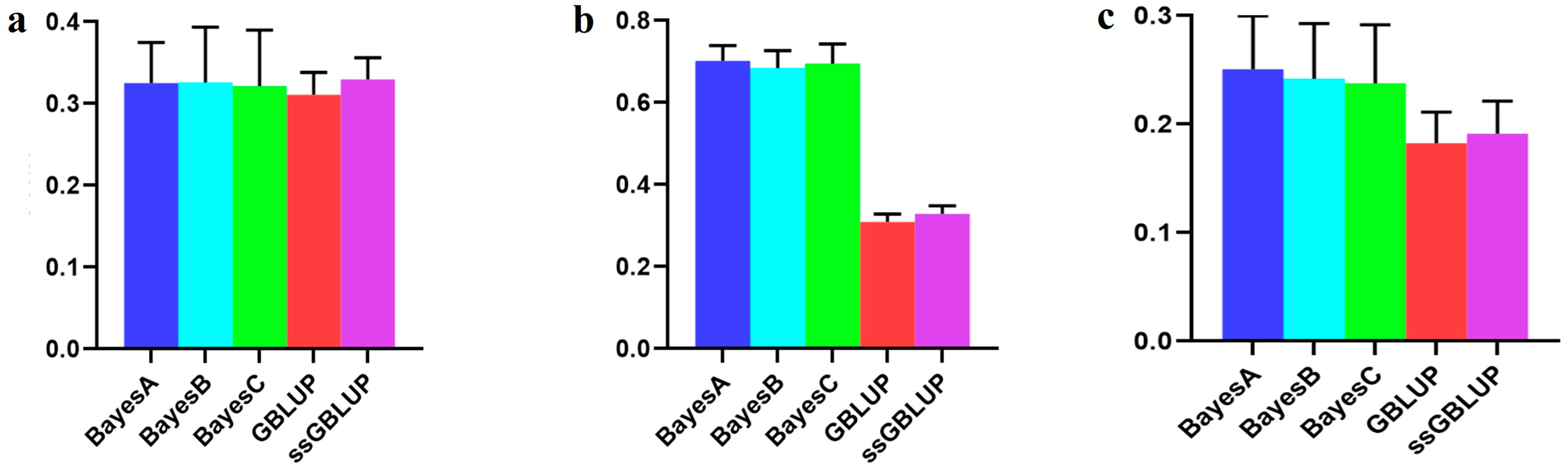
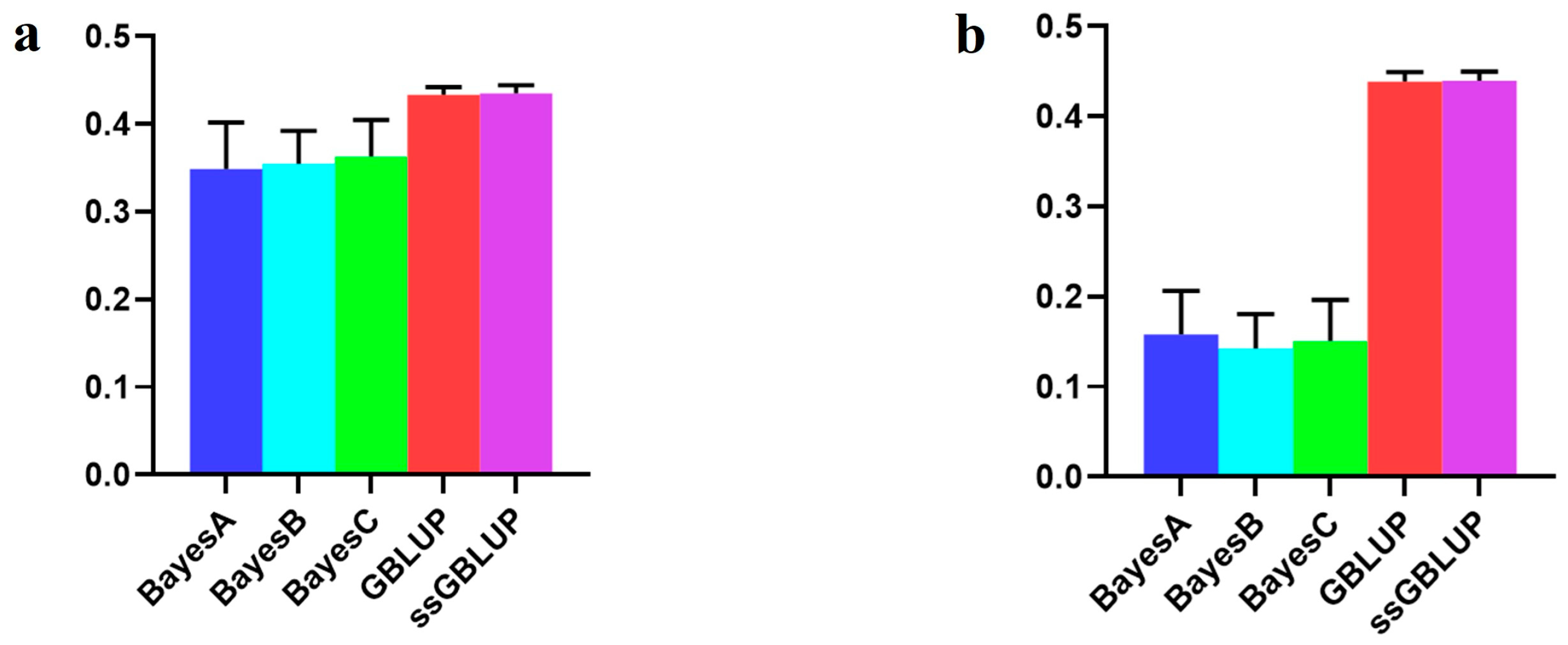
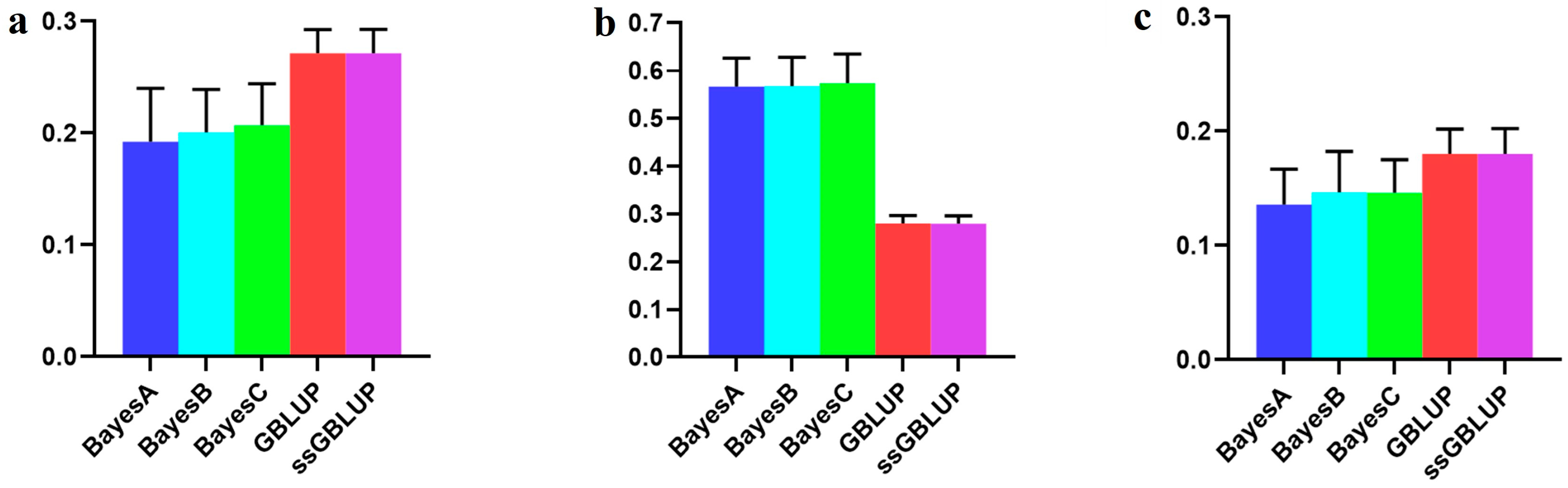
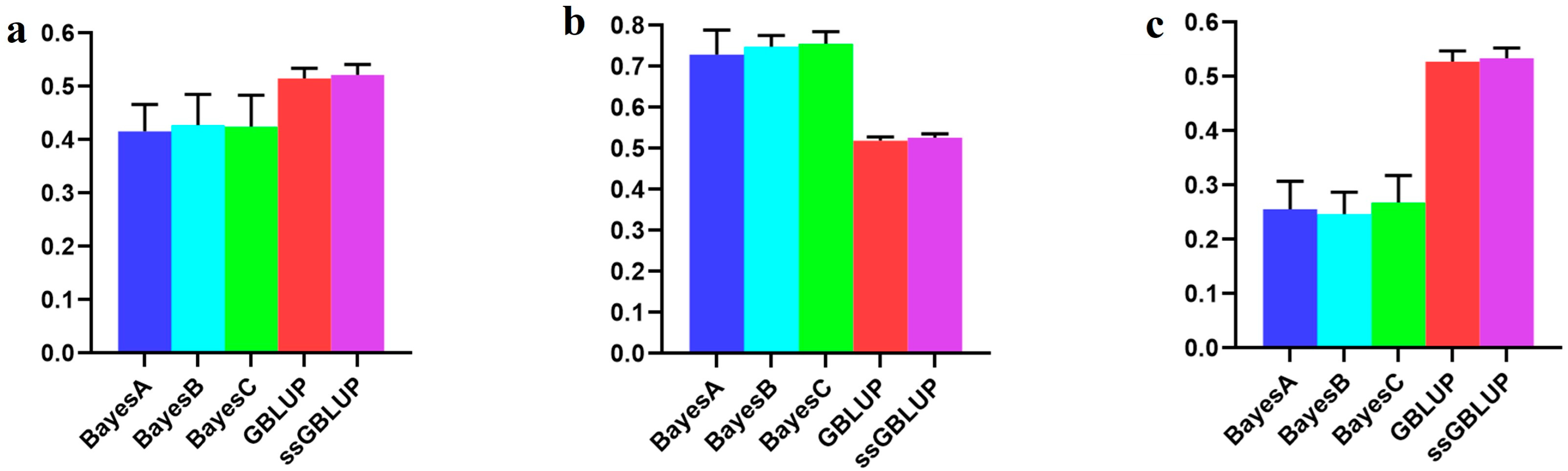
3.3. The Accuracy of the Canadian (Medium) Line
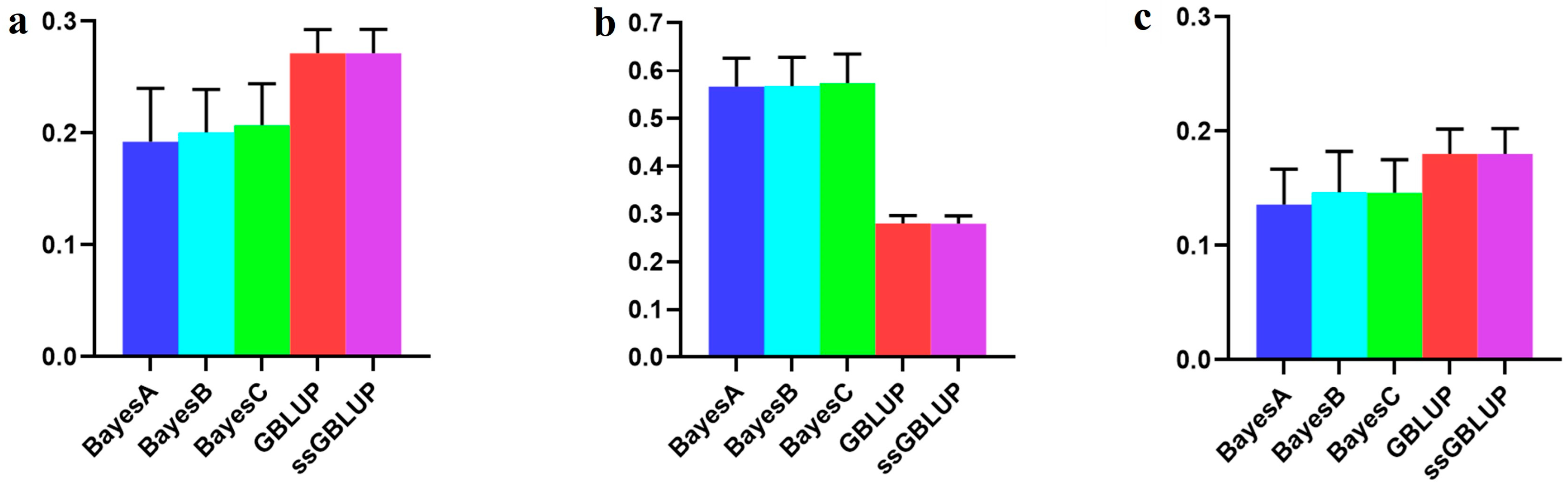
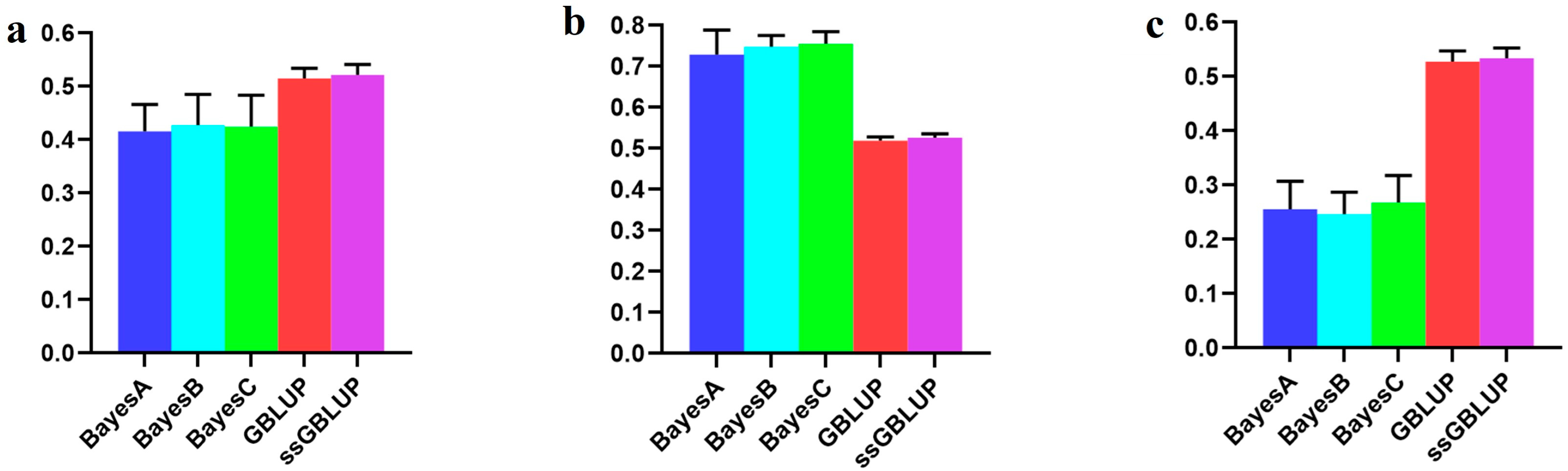
3.4. The Accuracy of the American (Large) Line
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de Los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.E.; Hayes, B.J. Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nat. Rev. Genet. 2009, 10, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.C.; Verbyla, K.; Goddard, M.E. Accuracy of genomic breeding values in multi-breed dairy cattle populations. Genet. Sel. Evol. 2009, 41, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Dekkers, J.C.M.; Fernando, R.L.; Jannink, J.L. Factors Affecting Accuracy From Genomic Selection in Populations Derived From Multiple Inbred Lines: A Barley Case Study. Genetics 2009, 182, 355–364. [Google Scholar] [CrossRef]
- Yin, C.; Zhou, P.; Wang, Y.; Yin, Z.; Liu, Y. Using genomic selection to improve the accuracy of genomic prediction for multi-populations in pigs. Anim. Int. J. Anim. Biosci. 2024, 18, 101062. [Google Scholar] [CrossRef]
- Guo, X.; Christensen, O.F.; Ostersen, T.; Wang, Y.; Lund, M.S.; Su, G. Improving genetic evaluation of litter size and piglet mortality for both genotyped and nongenotyped individuals using a single-step method. J. Anim. Sci. 2015, 93, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Wientjes, Y.C.; Veerkamp, R.F.; Bijma, P.; Bovenhuis, H.; Schrooten, C.; Calus, M.P. Empirical and deterministic accuracies of across-population genomic prediction. Genet. Sel. Evol. GSE 2015, 47, 5. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.B.; Nugent, R.A., 3rd. Heritability of body length and measures of body density and their relationship to backfat thickness and loin muscle area in swine. J. Anim. Sci. 2003, 81, 1943–1949. [Google Scholar] [CrossRef]
- Bidanel, J.P.; Ducos, A.; Gueblez, R.; Labroue, F. Genetic parameters of backfat thickness, age at 100 kg and ultimate pH in on-farm tested French Landrace and Large White pigs. Livest. Prod. Sci. 1994, 40, 291–301. [Google Scholar] [CrossRef]
- Davoli, R.; Catillo, G.; Serra, A.; Zappaterra, M.; Zambonelli, P.; Zilio, D.M.; Steri, R.; Mele, M.; Buttazzoni, L.; Russo, V. Genetic parameters of backfat fatty acids and carcass traits in Large White pigs. Anim. Int. J. Anim. Biosci. 2019, 13, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Wang, T. Genetic Parameter Estimates for the Age at 100 kg and Backfat Thickness in Large Whites. Master’s Thesis, China Agricultural University, Beijing, China, 2007. [Google Scholar]
- Chen, D.; Wu, P.; Yang, Q.; Wang, K.; Zhou, J.; Yang, X.; Jiang, A.; Shen, L.; Xiao, W.; Jiang, Y.; et al. Genome-wide association study for backfat thickness at 100 kg and loin muscle thickness in domestic pigs based on genotyping by sequencing. Physiol. Genom. 2019, 51, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef]
- Pérez, P.; de los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Soerensen, P.; Rohde, P.D.; Soerensen, I.F. qgg: Statistical Tools for Quantitative Genetic Analyses. R Package Version 1.1.2. Available online: https://github.com/psoerensen/qgg (accessed on 25 March 2023).
- Aguilar, I.; Misztal, I.; Johnson, D.; Legarra, A.; Tsuruta, S.; Lawlor, T. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, S.; Calus, M.P.; Meuwissen, T.H.; Wientjes, Y.C. Across population genomic prediction scenarios in which Bayesian variable selection outperforms GBLUP. BMC Genet. 2015, 16, 146. [Google Scholar] [CrossRef]
- Christensen, O.; Madsen, P.; Nielsen, B.; Ostersen, T.; Su, G. Single-step methods for genomic evaluation in pigs. Animal 2012, 6, 1565–1571. [Google Scholar] [CrossRef]
- Gao, H.; Christensen, O.F.; Madsen, P.; Nielsen, U.S.; Zhang, Y.; Lund, M.S.; Su, G. Comparison on genomic predictions using three GBLUP methods and two single-step blending methods in the Nordic Holstein population. Genet. Sel. Evol. GSE 2012, 44, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, S.; Huang, J.; Li, L.; Zhang, Q.; Ding, X. Improving the accuracy of genomic prediction in Chinese Holstein cattle by using one-step blending. Genet. Sel. Evol. 2014, 46, 66. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Chang, H.K.; Lee, S.S.; Choi, T.J. Genetic Analysis of Major Production and Reproduction Traits of Korean Duroc, Landrace and Yorkshire Pigs. Animals 2021, 11, 1321. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.I.; Lee, S.-H.; Shin, D.-H.; Oh, J.-D.; Chai, H.-H.; Park, W.; Park, J.-E.; Lim, D. Accuracy of genomic evaluation using imputed high-density genotypes for carcass traits in commercial Hanwoo population. Livest. Sci. 2020, 241, 104256. [Google Scholar] [CrossRef]
- Song, H.; Zhang, J.; Jiang, Y.; Gao, H.; Tang, S.; Mi, S.; Yu, F.; Meng, Q.; Xiao, W.; Zhang, Q.; et al. Genomic prediction for growth and reproduction traits in pig using an admixed reference population. J. Anim. Sci. 2017, 95, 3415–3424. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, Z.; Ma, P.; Wang, Z.; Wang, Q.; Zhang, Z.; Pan, Y. The effect of high-density genotypic data and different methods on joint genomic prediction: A case study in large white pigs. Anim. Genet. 2023, 54, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Cappa, E.P.; de Lima, B.M.; da Silva-Junior, O.B.; Garcia, C.C.; Mansfield, S.D.; Grattapaglia, D. Improving genomic prediction of growth and wood traits in Eucalyptus using phenotypes from non-genotyped trees by single-step GBLUP. Plant Sci. Int. J. Exp. Plant Biol. 2019, 284, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Misztal, I.; Lourenco, D.; Legarra, A. Current status of genomic evaluation. J. Anim. Sci. 2020, 98, skaa101. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G.; Weir, B.S. Variation in actual relationship as a consequence of Mendelian sampling and linkage. Genet. Res. 2011, 93, 47–64. [Google Scholar] [CrossRef]
- Lund, M.S.; de Roos, A.P.; de Vries, A.G.; Druet, T.; Ducrocq, V.; Fritz, S.; Guillaume, F.; Guldbrandtsen, B.; Liu, Z.; Reents, R. A common reference population from four European Holstein populations increases reliability of genomic predictions. Genet. Sel. Evol. 2011, 43, 43. [Google Scholar] [CrossRef]
- Weber, B.; Hu, S.X.; Steiner, M.; Zhao, F.C. A diverse ichnofauna from the Cambrian Stage 4 Wulongqing Formation near Kunming (Yunnan Province, South China). Bull. Geosci. 2012, 87, 71–92. [Google Scholar] [CrossRef]
- van den Berg, I.; Meuwissen, T.H.E.; MacLeod, I.M.; Goddard, M.E. Predicting the effect of reference population on the accuracy of within, across, and multibreed genomic prediction. J. Dairy Sci. 2019, 102, 3155–3174. [Google Scholar] [CrossRef] [PubMed]
- Karaman, E.; Su, G.; Croue, I.; Lund, M.S. Genomic prediction using a reference population of multiple pure breeds and admixed individuals. Genet. Sel. Evol. GSE 2021, 53, 46. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Li, J.; Zhang, Z. Multi-omics-data-assisted genomic feature markers preselection improves the accuracy of genomic prediction. J. Anim. Sci. Biotechnol. 2020, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- Cuyabano, B.C.D.; Su, G.; Lund, M.S. Selection of haplotype variables from a high-density marker map for genomic prediction. Genet. Sel. Evol. 2015, 47, 61. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.; Druet, T.; Hess, A.; Garrick, D. Fixed-length haplotypes can improve genomic prediction accuracy in an admixed dairy cattle population. Genet. Sel. Evol. 2017, 49, 54. [Google Scholar] [CrossRef]
- Ye, H.; Zhang, Z.; Ren, D.; Cai, X.; Zhu, Q.; Ding, X.; Zhang, H.; Zhang, Z.; Li, J. Genomic Prediction Using LD-Based Haplotypes in Combined Pig Populations. Front. Genet. 2022, 13, 843300. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scenarios | BayesA | BayesB | BayesC | GBLUP | ssGBLUP |
|---|---|---|---|---|---|
| Combined American, Canadian, and Danish populations | 0.10850 | 0.10687 | 0.10595 | 0.13385 | 0.14336 |
| Combined American and Canadian populations | 0.65999 | 0.66093 | 0.64634 | 0.11604 | 0.12715 |
| Danish populations individually | 0.04895 | 0.04387 | 0.04385 | 0.05784 | 0.05588 |
| Scenarios | BayesA | BayesB | BayesC | GBLUP | ssGBLUP |
|---|---|---|---|---|---|
| Combined American, Canadian, and Danish populations | 0.32526 | 0.32599 | 0.32159 | 0.31096 | 0.32953 |
| Combined American and Canadian populations | 0.70134 | 0.68404 | 0.69489 | 0.30901 | 0.32876 |
| Danish populations individually | 0.25057 | 0.24194 | 0.23761 | 0.18266 | 0.19131 |
| Scenarios | BayesA | BayesB | BayesC | GBLUP | ssGBLUP |
|---|---|---|---|---|---|
| Combined American, Canadian, and Danish populations | 0.19236 | 0.20061 | 0.20714 | 0.27130 | 0.27133 |
| Combined American and Danish populations | 0.56656 | 0.56813 | 0.57448 | 0.28029 | 0.27973 |
| Canadian population individually | 0.13579 | 0.14652 | 0.14622 | 0.18004 | 0.18012 |
| Scenarios | BayesA | BayesB | BayesC | GBLUP | ssGBLUP |
|---|---|---|---|---|---|
| Combined American, Canadian, and Danish | 0.41599 | 0.42777 | 0.42452 | 0.51505 | 0.52175 |
| Combined American and Danish populations | 0.72887 | 0.74858 | 0.75569 | 0.51873 | 0.52609 |
| Canadian population individually | 0.25572 | 0.24653 | 0.26800 | 0.52769 | 0.53372 |
| Scenarios | BayesA | BayesB | BayesC | GBLUP | ssGBLUP |
|---|---|---|---|---|---|
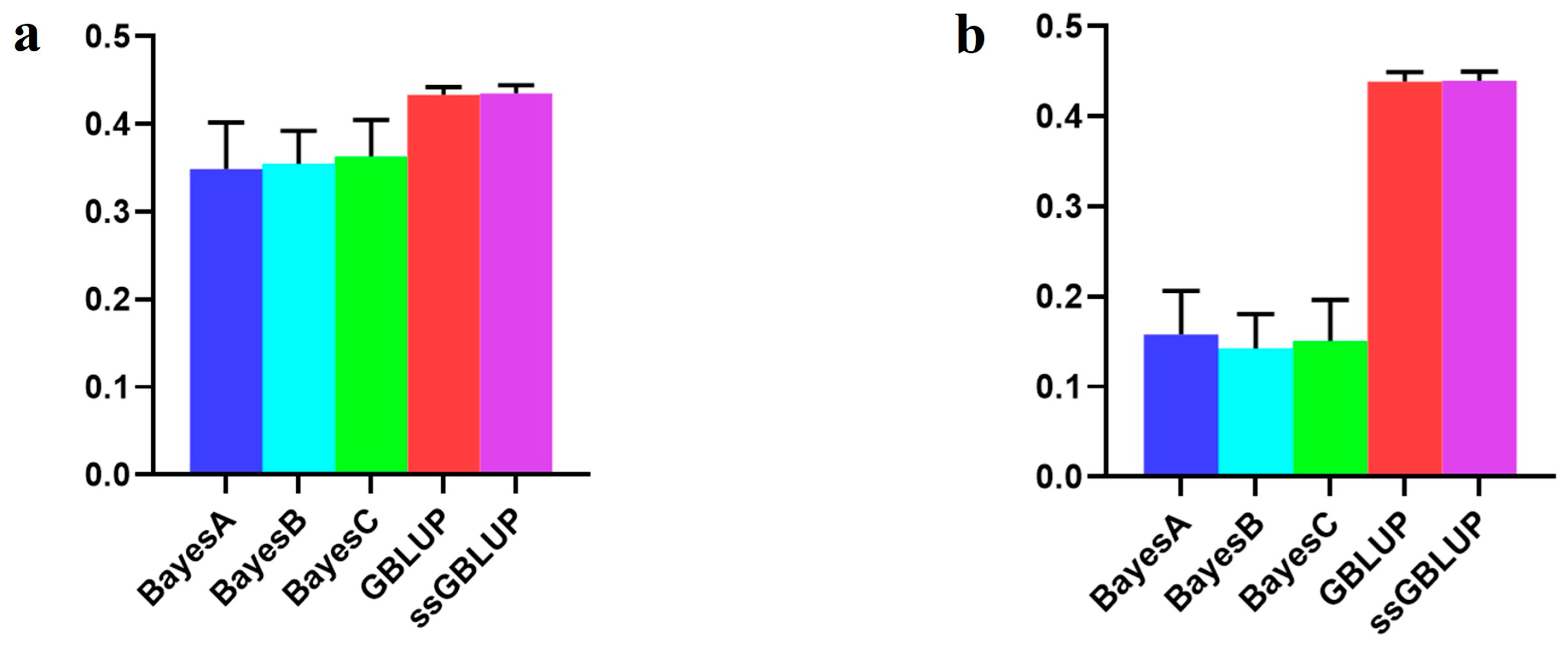
| Combined American, Canadian, and Danish populations | 0.34910 | 0.35499 | 0.36329 | 0.43341 | 0.43526 |
| American population individually | 0.15793 | 0.14229 | 0.15109 | 0.43869 | 0.43978 |
| Scenarios | BayesA | BayesB | BayesC | GBLUP | ssGBLUP |
|---|---|---|---|---|---|
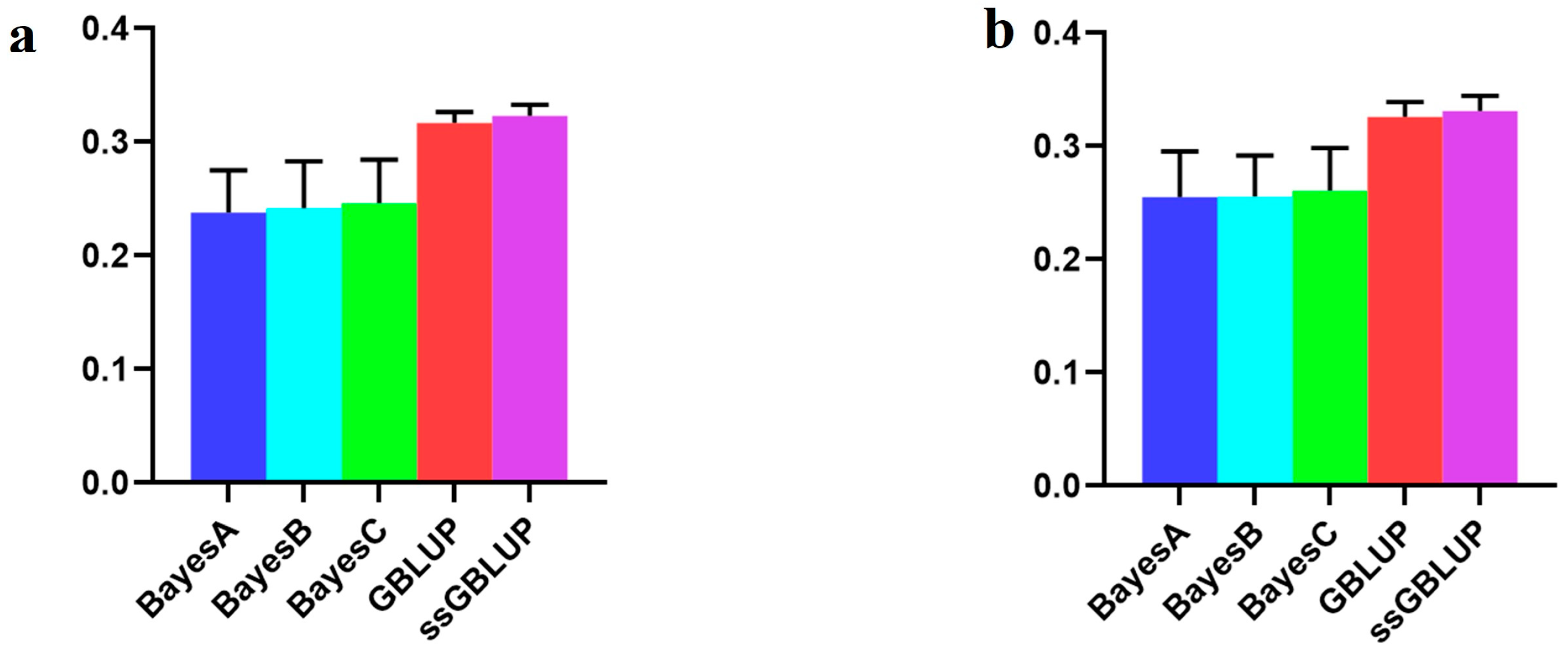
| Combined American, Canadian, and Danish populations | 0.23780 | 0.24182 | 0.24619 | 0.31689 | 0.32331 |
| American population individually | 0.25502 | 0.25556 | 0.26096 | 0.32570 | 0.33130 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, C.; Shi, H.; Zhou, P.; Wang, Y.; Tao, X.; Yin, Z.; Zhang, X.; Liu, Y. Genomic Prediction of Growth Traits in Yorkshire Pigs of Different Reference Group Sizes Using Different Estimated Breeding Value Models. Animals 2024, 14, 1098. https://doi.org/10.3390/ani14071098
Yin C, Shi H, Zhou P, Wang Y, Tao X, Yin Z, Zhang X, Liu Y. Genomic Prediction of Growth Traits in Yorkshire Pigs of Different Reference Group Sizes Using Different Estimated Breeding Value Models. Animals. 2024; 14(7):1098. https://doi.org/10.3390/ani14071098
Chicago/Turabian StyleYin, Chang, Haoran Shi, Peng Zhou, Yuwei Wang, Xuzhe Tao, Zongjun Yin, Xiaodong Zhang, and Yang Liu. 2024. "Genomic Prediction of Growth Traits in Yorkshire Pigs of Different Reference Group Sizes Using Different Estimated Breeding Value Models" Animals 14, no. 7: 1098. https://doi.org/10.3390/ani14071098
APA StyleYin, C., Shi, H., Zhou, P., Wang, Y., Tao, X., Yin, Z., Zhang, X., & Liu, Y. (2024). Genomic Prediction of Growth Traits in Yorkshire Pigs of Different Reference Group Sizes Using Different Estimated Breeding Value Models. Animals, 14(7), 1098. https://doi.org/10.3390/ani14071098