
Surveillance and Genetic Analysis of Low-Pathogenicity Avian Influenza Viruses Isolated from Feces of Wild Birds in Mongolia, 2021 to 2023

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Molecular Detection and Virus Isolation
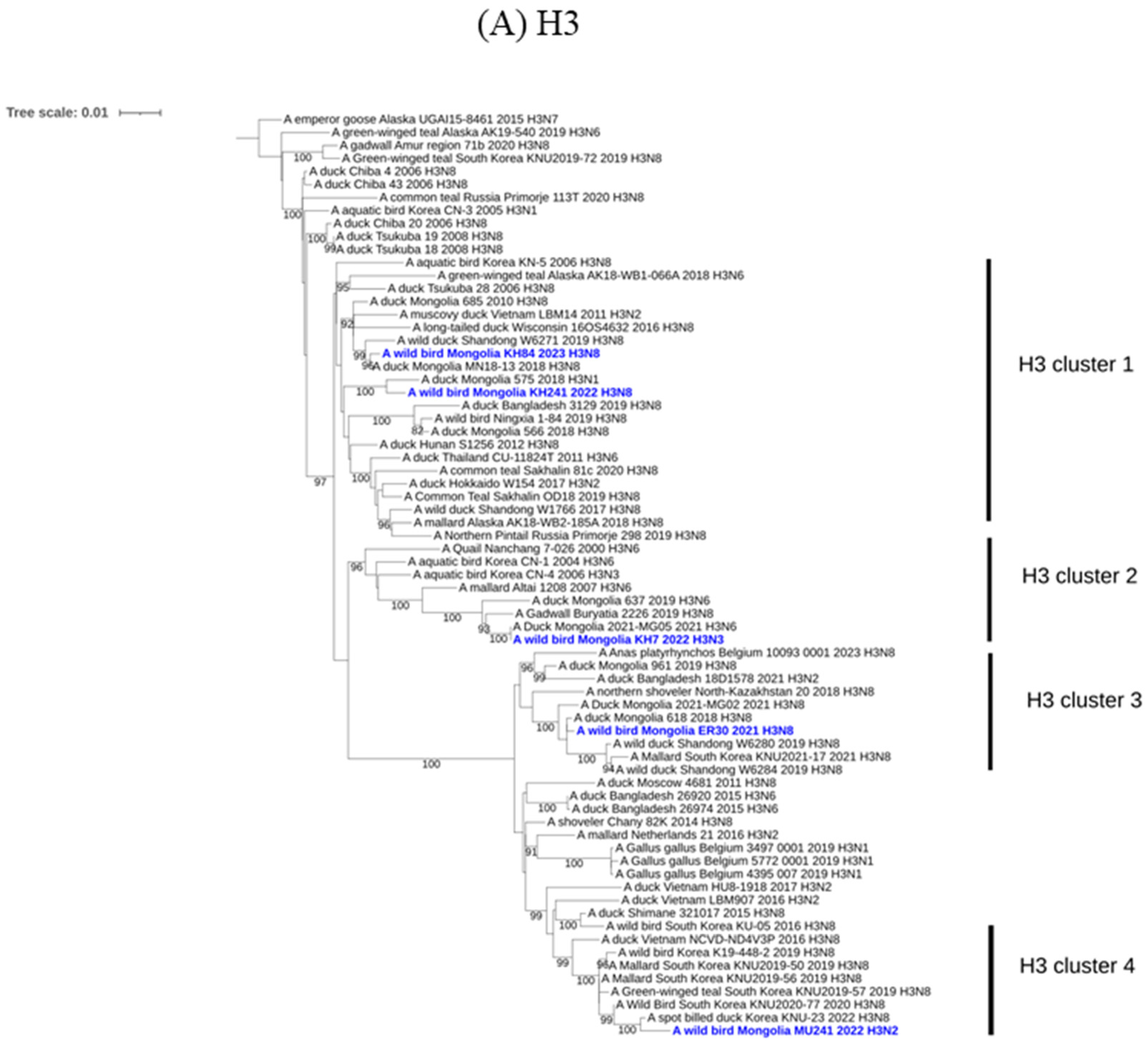
2.3. Molecular and Phylogenetic Analysis
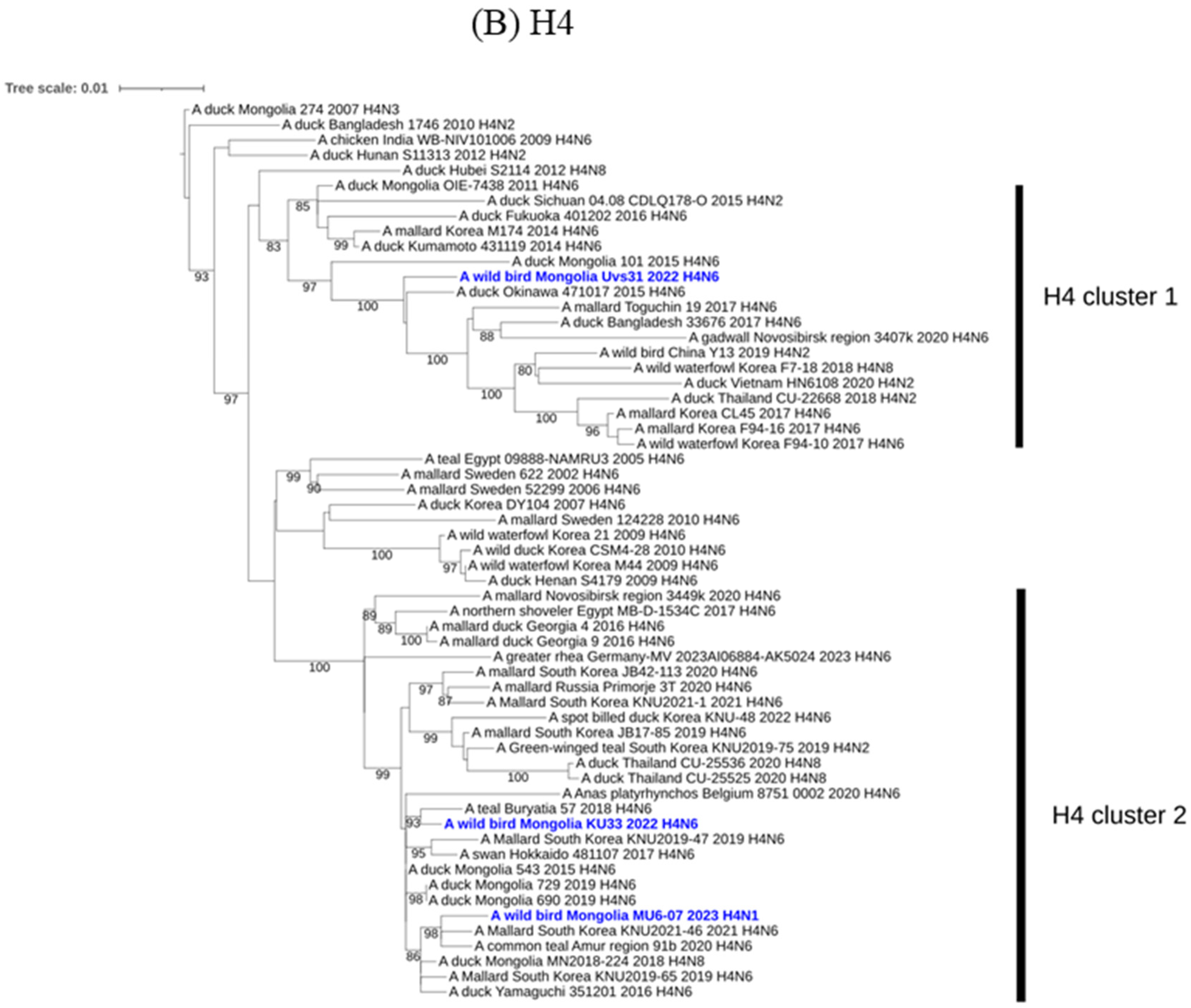
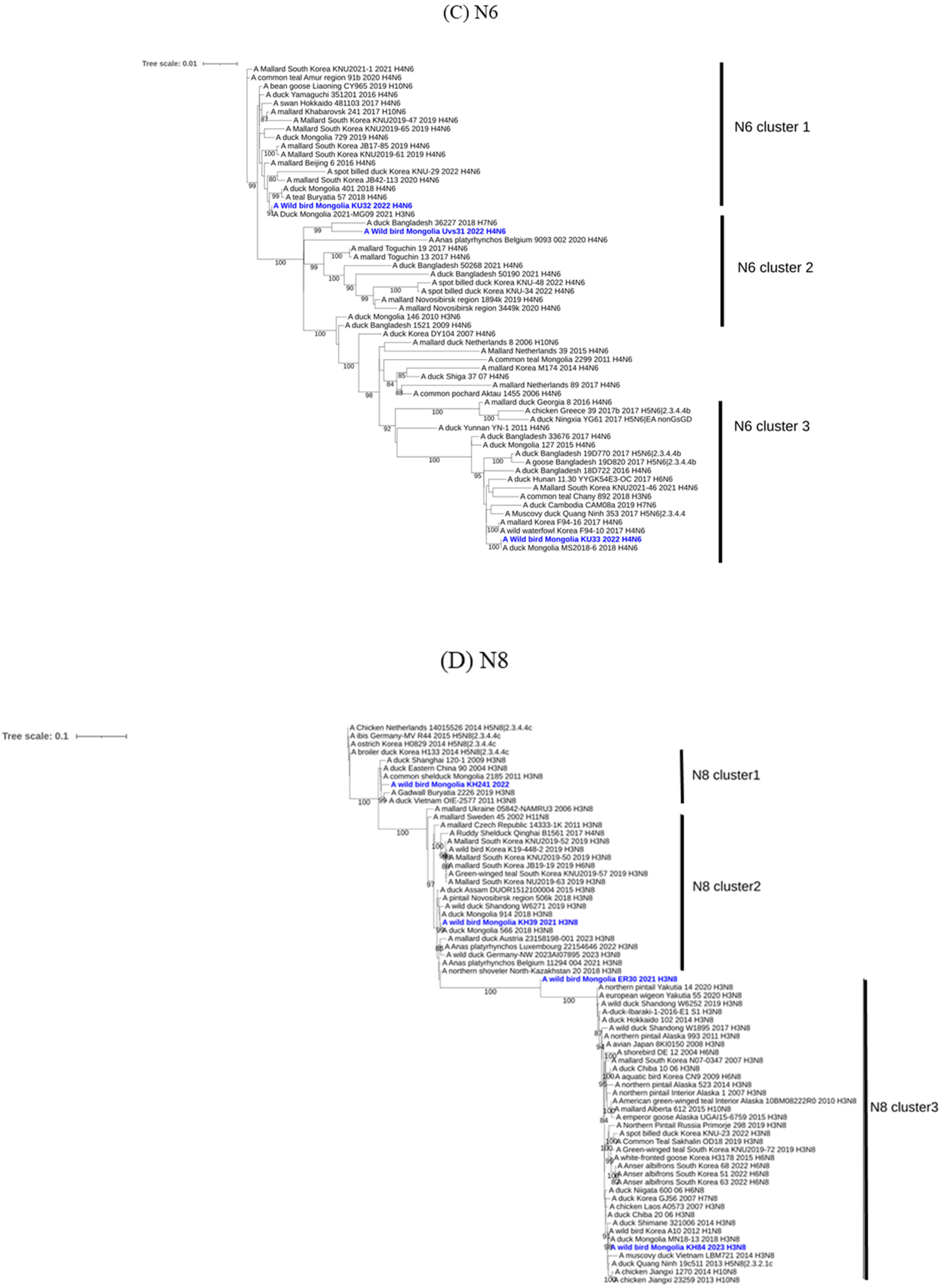
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krauss, S.; Webster, R.G. Avian influenza virus surveillance and wild birds: Past and present. Avian Dis. 2010, 54 (Suppl. S1), 394–398. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.M.; Lee, C.W.; Gourapura, R.; Saif, Y.M. Interspecies and intraspecies transmission of influenza A viruses: Viral, host and environmental factors. Anim. Health Res. Rev. 2010, 11, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Moorman, J.P. Viral characteristics of influenza. South. Med. J. 2003, 96, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Li, X.; Goraya, M.U.; Wang, S.; Chen, J. Evolution of Influenza A Virus by Mutation and Re-Assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Smith, G.J.D.; Zhang, S.Y.; Qin, J.; Li, K.S.; Webster, R.G.; Peiris, J.S.M.; Guan, Y. Avian flu: H5N1 virus outbreak in migratory waterfowl. Nature 2005, 436, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Pasick, J.; Berhane, Y.; Joseph, T.; Bowes, V.; Hisanaga, T.; Handel, K.; Alexandersen, S. Reassortant highly pathogenic influenza A H5N2 virus containing gene segments related to Eurasian H5N8 in British Columbia, Canada, 2014. Sci. Rep. 2015, 5, 9484. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, M.D.; Artois, J.; Dellicour, S.; Lemey, P.; Dauphin, G.; Dobschuetz, S.V.; Boeckel, T.P.V.; Castellan, D.M.; Morzaria, S.; Gilbert, M. Geographical and Historical Patterns in the Emergences of Novel Highly Pathogenic Avian Influenza (HPAI) H5 and H7 Viruses in Poultry. Front. Vet. Sci. 2018, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Lp, H.S.; Torchetti, M.K.; Crespo, R.; Kohrs, P.; DeBruyn, P.; Masnfield, K.G.; Baszler, T.; Badcoe, L.; Bodenstein, B.; Shearn-Bochsler, V.; et al. Novel Eurasian highly pathogenic avian influenza A H5 viruses in wild birds, Washington, USA, 2014. Emerg. Infect. Dis. 2015, 21, 886–890. [Google Scholar]
- Gombobaatar, S.; Samiya, D.; Baillie, J.M. Bird Red List and Its Future Development in Mongolia. Erforsch. Biol. Ressourcen Der Mong. 2012, 12, 169–182. [Google Scholar]
- López-Guerrero, P.; Maridet, O.; Zhang, Z.; Daxner-Höck, G. A new species of Argyromys (Rodentia, Mammalia) from the Oligocene of the Valley of Lakes (Mongolia): Its importance for palaeobiogeographical homogeneity across Mongolia, China and Kazakhstan. PLoS ONE 2017, 12, e0172733. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, S.; Morimoto, G.; Kitamura, W.; Imanishi, S.; Azuma, N. Rice fields along the East Asian-Australasian flyway are important habitats for an inland wader’s migration. Sci. Rep. 2020, 10, 4118. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.R.; Cho, A.Y.; Si, Y.J.; Lee, S.I.; Kim, D.J.; Jeong, H.S.; Kwon, J.H.; Song, C.S.; Lee, D.H. Evolution and Spread of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus in Wild Birds, South Korea, 2022–2023. Emerg. Infect. Dis. 2024, 30, 299–309. [Google Scholar] [CrossRef]
- Lycett, S.J.; Pohlmann, A.; Staubach, C.; Caliendo, V.; Woolhouse, M.; Beer, M.; Kuiken, T. Genesis and spread of multiple reassortants during the 2016/2017 H5 avian influenza epidemic in Eurasia. Proc. Natl. Acad. Sci. USA 2020, 117, 20814–20825. [Google Scholar] [CrossRef] [PubMed]
- Batbayar, N.; Takekawa, J.Y.; Newman, S.H.; Prosser, D.J.; NAtsagdorj, T.; Xiao, X. Migration strategies of Swan Geese Anser cygnoides from northeast Mongolia. Wlidfowl 2011, 61, 90–109. [Google Scholar]
- Ulaankhuu, A.; Bazarragchaa, E.; Okamatsu, M.; Hiono, T.; Bodisaikhan, K.; Amartuvshin, T.; Tserenjave, J.; Urangoo, T.; Buyantogtokh, K.; Matsuno, K.; et al. Genetic and antigenic characterization of H5 and H7 avian influenza viruses isolated from migratory waterfowl in Mongolia from 2017 to 2019. Virus Genes 2020, 56, 472–479. [Google Scholar] [CrossRef]
- Kang, H.M.; Kim, M.C.; Choi, J.G.; Batchuluun, D.; Erdene-Ochir, T.O.; Paek, M.R.; Sodnomdarjaa, R.; Kwon, J.H.; Lee, Y.J. Genetic analyses of avian influenza viruses in Mongolia, 2007 to 2009, and their relationships with Korean isolates from domestic poultry and wild birds. Poult. Sci. 2011, 90, 2229–2242. [Google Scholar] [CrossRef]
- Barkhasbaatar, A.; Gilbert, M.; Fine, A.E.; Shiilegdamba, E.; Damdinjav, B.; Buuveibaatar, B.; Khishgee, B.; Johnson, C.K.; Leung, C.Y.H.; Ankhanbaatar, U.; et al. Ecological characterization of 175 low-pathogenicity avian influenza viruses isolated from wild birds in Mongolia, 2009–2013 and 2016–2018. Vet. Med. Sci. 2023, 9, 2676–2685. [Google Scholar] [CrossRef]
- Jeong, S.; Otgontogtokh, N.; Lee, D.H.; Davganyam, B.; Lee, S.H.; Cho, A.Y.; Tseren-Ochir, E.O.; Song, C.S. Highly Pathogenic Avian Influenza Clade 2.3.4.4 Subtype H5N6 Viruses Isolated from Wild Whooper Swans, Mongolia, 2020. Emerg. Infect. Dis. 2021, 27, 1181–1183. [Google Scholar] [CrossRef]
- Kim, G.S.; Kim, T.S.; Son, J.S.; Lai, V.D.; Park, J.E.; Wang, S.J.; Jheong, W.H.; Mo, I.P. The difference of detection rate of avian influenza virus in the wild bird surveillance using various methods. J. Vet. Sci. 2019, 20, e56. [Google Scholar] [CrossRef]
- Lee, D.H. Complete Genome Sequencing of Influenza A Viruses Using Next-Generation Sequencing. Methods Mol. Biol. 2020, 2123, 69–79. [Google Scholar] [PubMed]
- Miller, M.A.; Terri Schwartz, W.T.P. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, C.; Sun, H.T.; Ma, J.; Qi, Y.; Qin, S.Y. Prevalence of avian influenza viruses and their associated antibodies in wild birds in China: A systematic review and meta-analysis. Microb. Pathog. 2019, 135, 103613. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.; Ashton-Butt, A.; Scott, G.; Reid, S.M.; Coward, V.; Hansen, R.D.E.; Banyard, A.C.; Ward, A.I. High pathogenicity avian influenza: Targeted active surveillance of wild birds to enable early detection of emerging disease threats. Epidemiol. Infect. 2022, 151, e15. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhan, D.; Li, L.; Lei, F.; Liu, B.; Liu, D.; Xiao, H.; Feng, Y.; Li, J.; Yang, B.; et al. H5N1 avian influenza re-emergence of Lake Qinghai: Phylogenetic and antigenic analyses of the newly isolated viruses and roles of migratory birds in virus circulation. J. Gen. Virol. 2008, 89 Pt 3, 697–702. [Google Scholar]
- Cui, P.; Shi, J.; Wang, C.; Zhang, Y.; Xing, X.; Kong, H.; Yan, C.; Zeng, X.; Liu, L.; Tian, G.; et al. Global dissemination of H5N1 influenza viruses bearing the clade 2.3.4.4b HA gene and biologic analysis of the ones detected in China. Emerg. Microbes Infect. 2022, 11, 1693–1704. [Google Scholar] [CrossRef]
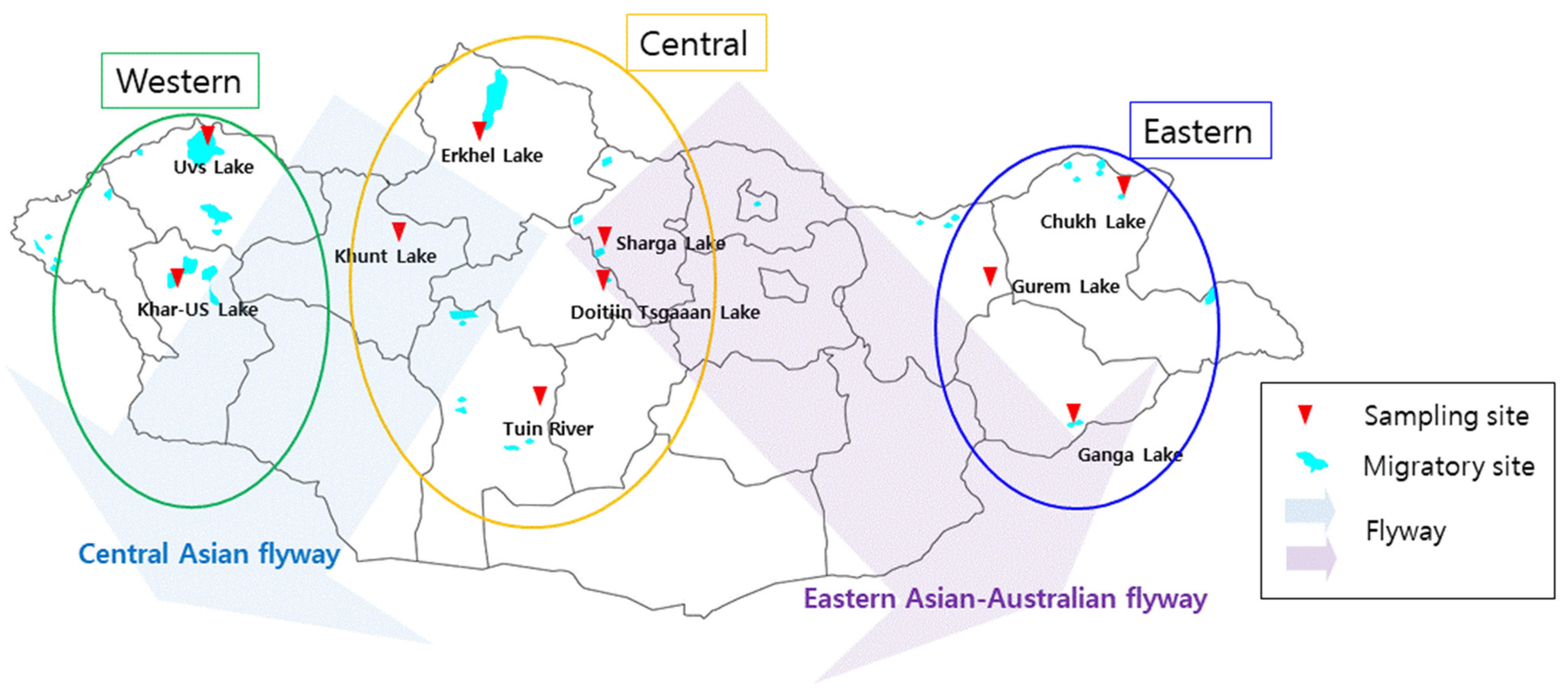



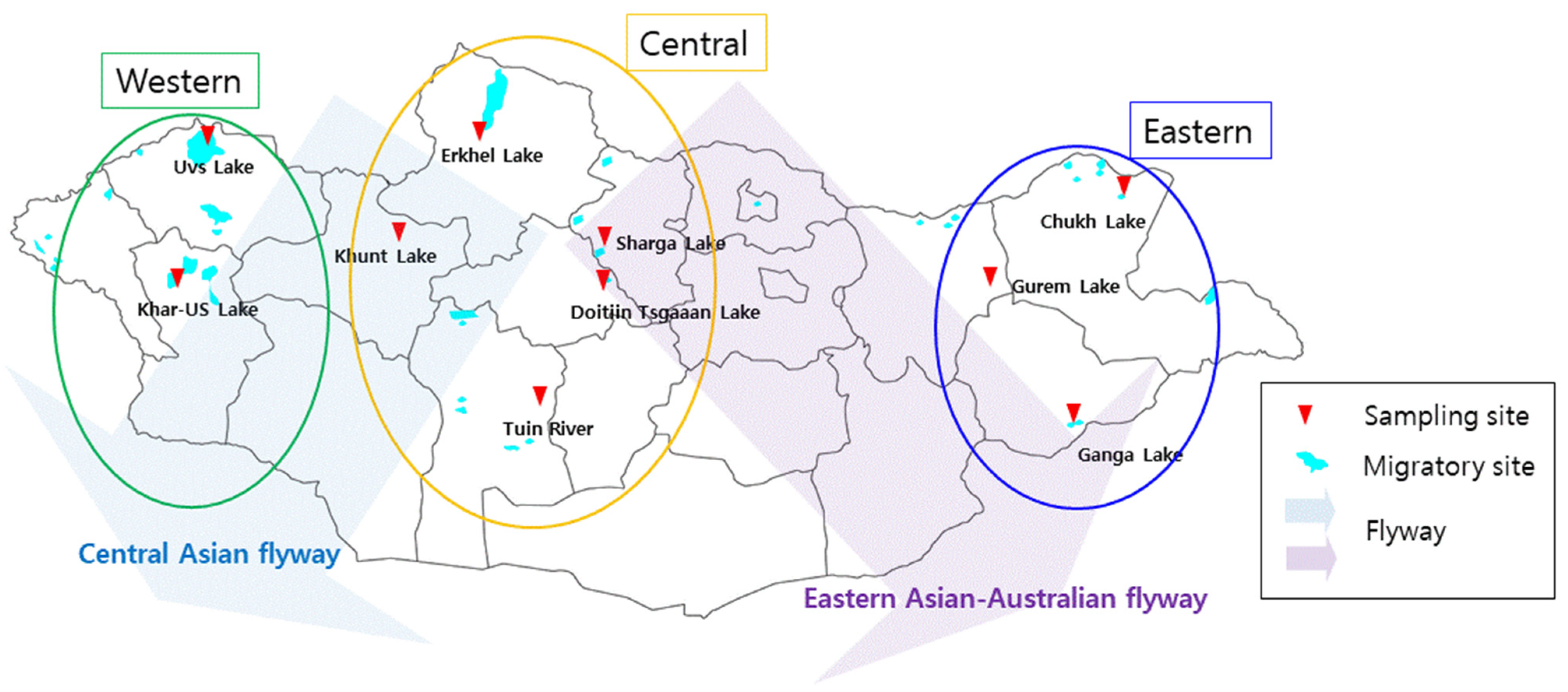{kind=link}
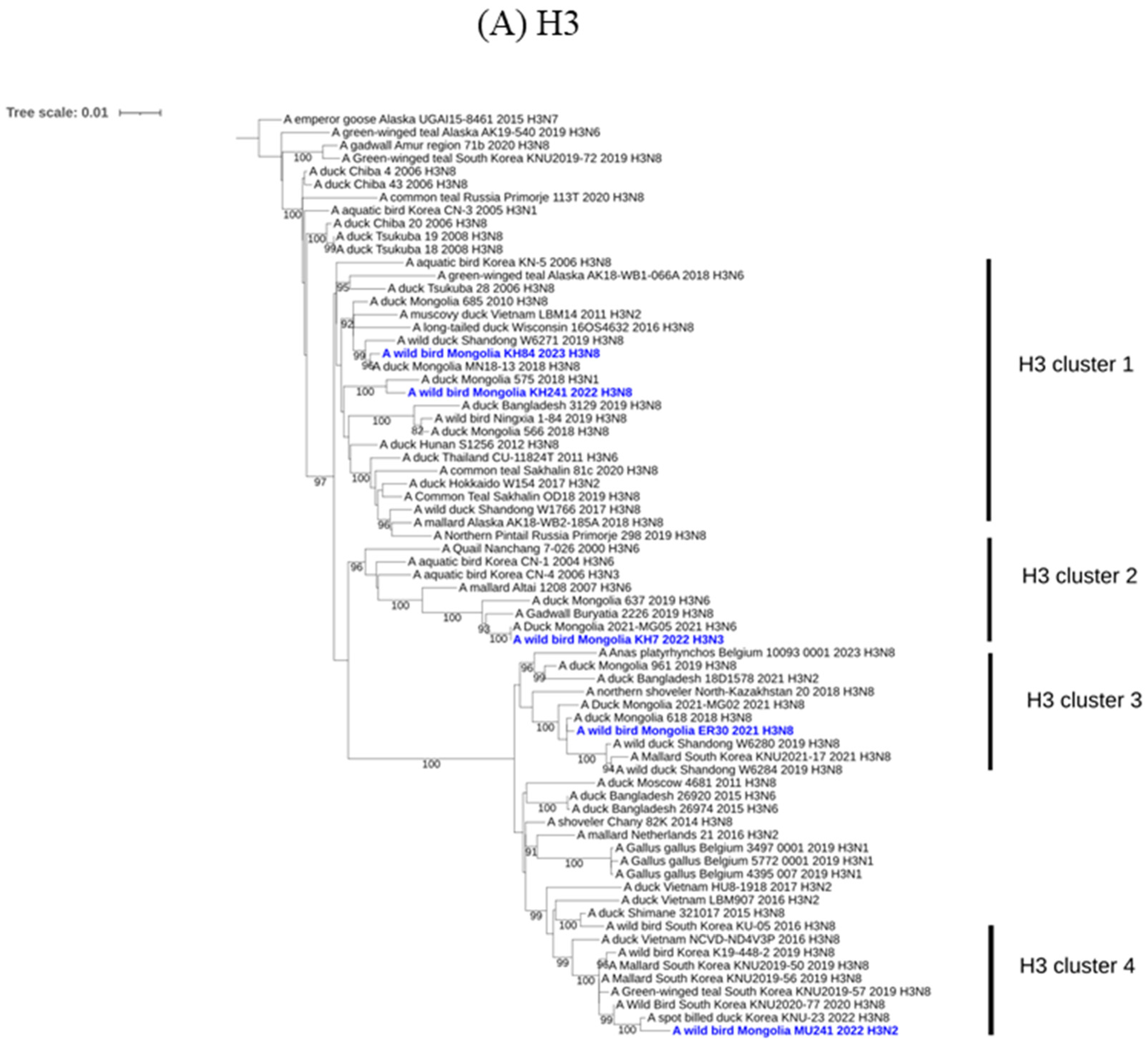{kind=link}
{kind=link}
{kind=link}
| Region of Collection | Total Numbers of Samples | M-Positive Samples | Virus Isolation | ||||
|---|---|---|---|---|---|---|---|
| Year | Western (1) | Central (2) | Eastern (3) | No. | % (4) | ||
| 2021 | - | 2812 | 1334 | 4146 | 39 | 0.94 | 30 |
| 2022 | 702 | 794 | 641 | 2137 | 33 | 1.54 | 24 |
| 2023 | 1094 | 1385 | 1387 | 3866 | 31 | 0.80 | 23 |
| Total | 1796 | 4991 | 3362 | 10,149 | 103 | 1.01 | 77 |
| Subtype | 2021 | 2022 | 2023 | Total |
|---|---|---|---|---|
| H1N2 | 0 | 1 | 1 | 2 |
| H2N8 | 0 | 2 | 0 | 1 |
| H3N1 | 0 | 1 | 0 | 1 |
| H3N2 | 0 | 2 | 3 | 5 |
| H3N3 | 4 | 2 | 0 | 6 |
| H3N8 | 19 | 5 | 6 | 30 |
| H4N1 | 0 | 0 | 3 | 7 |
| H4N6 | 7 | 4 | 4 | 11 |
| H6N1 | 0 | 4 | 2 | 6 |
| H6N8 | 0 | 1 | 0 | 1 |
| H10N7 | 0 | 2 | 0 | 2 |
| H10N8 | 0 | 0 | 3 | 3 |
| H13N8 | 0 | 0 | 1 | 1 |
| Total | 30 | 24 | 23 | 77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, Y.-M.; Tseren Ochir, E.-O.; Heo, G.-B.; An, S.-H.; Jeong, H.; Dondog, U.; Myagmarsuren, T.; Lee, Y.-J.; Lee, K.-N. Surveillance and Genetic Analysis of Low-Pathogenicity Avian Influenza Viruses Isolated from Feces of Wild Birds in Mongolia, 2021 to 2023. Animals 2024, 14, 1105. https://doi.org/10.3390/ani14071105
Kang Y-M, Tseren Ochir E-O, Heo G-B, An S-H, Jeong H, Dondog U, Myagmarsuren T, Lee Y-J, Lee K-N. Surveillance and Genetic Analysis of Low-Pathogenicity Avian Influenza Viruses Isolated from Feces of Wild Birds in Mongolia, 2021 to 2023. Animals. 2024; 14(7):1105. https://doi.org/10.3390/ani14071105
Chicago/Turabian StyleKang, Yong-Myung, Erdene-Ochir Tseren Ochir, Gyeong-Beom Heo, Se-Hee An, Hwanseok Jeong, Urankhaich Dondog, Temuulen Myagmarsuren, Youn-Jeong Lee, and Kwang-Nyeong Lee. 2024. "Surveillance and Genetic Analysis of Low-Pathogenicity Avian Influenza Viruses Isolated from Feces of Wild Birds in Mongolia, 2021 to 2023" Animals 14, no. 7: 1105. https://doi.org/10.3390/ani14071105