
Comparative Analysis of How the Fecal Microbiota of Green-Winged Saltator (Saltator similis) Diverge among Animals Living in Captivity and in Wild Habitats
 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
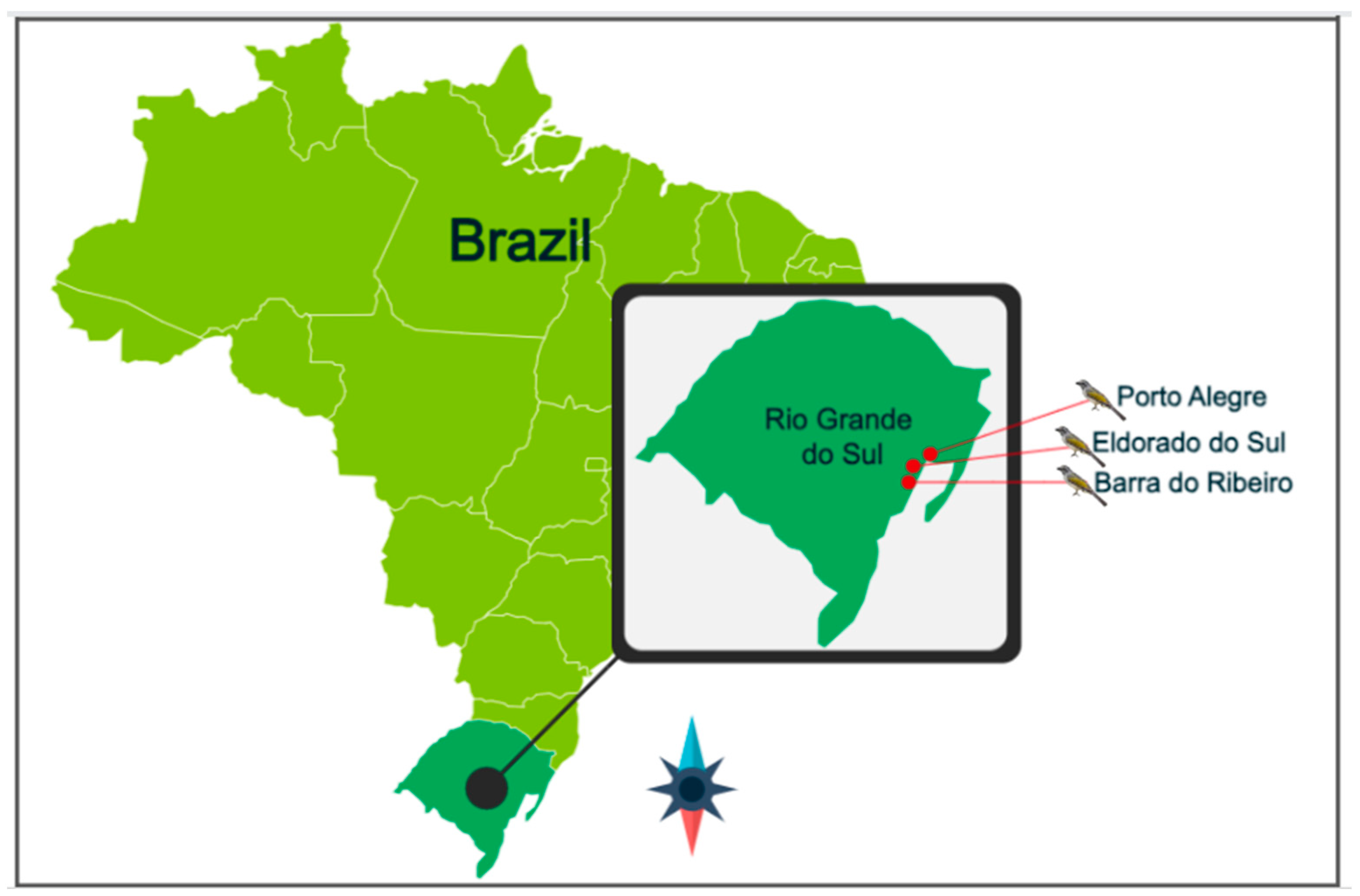
2.1. Animal Selection, Capture, and Collection of Fecal Samples
2.2. Extraction of Nucleic Acids of Fecal Samples from Saltator Similis
2.3. Amplification and Sequencing of the Bacterial Community of Fecal Samples
2.4. Taxonomic Analysis of the Bacterial Community from Fecal Samples
2.5. Statistical Analyses of the Taxonomic Results
3. Results
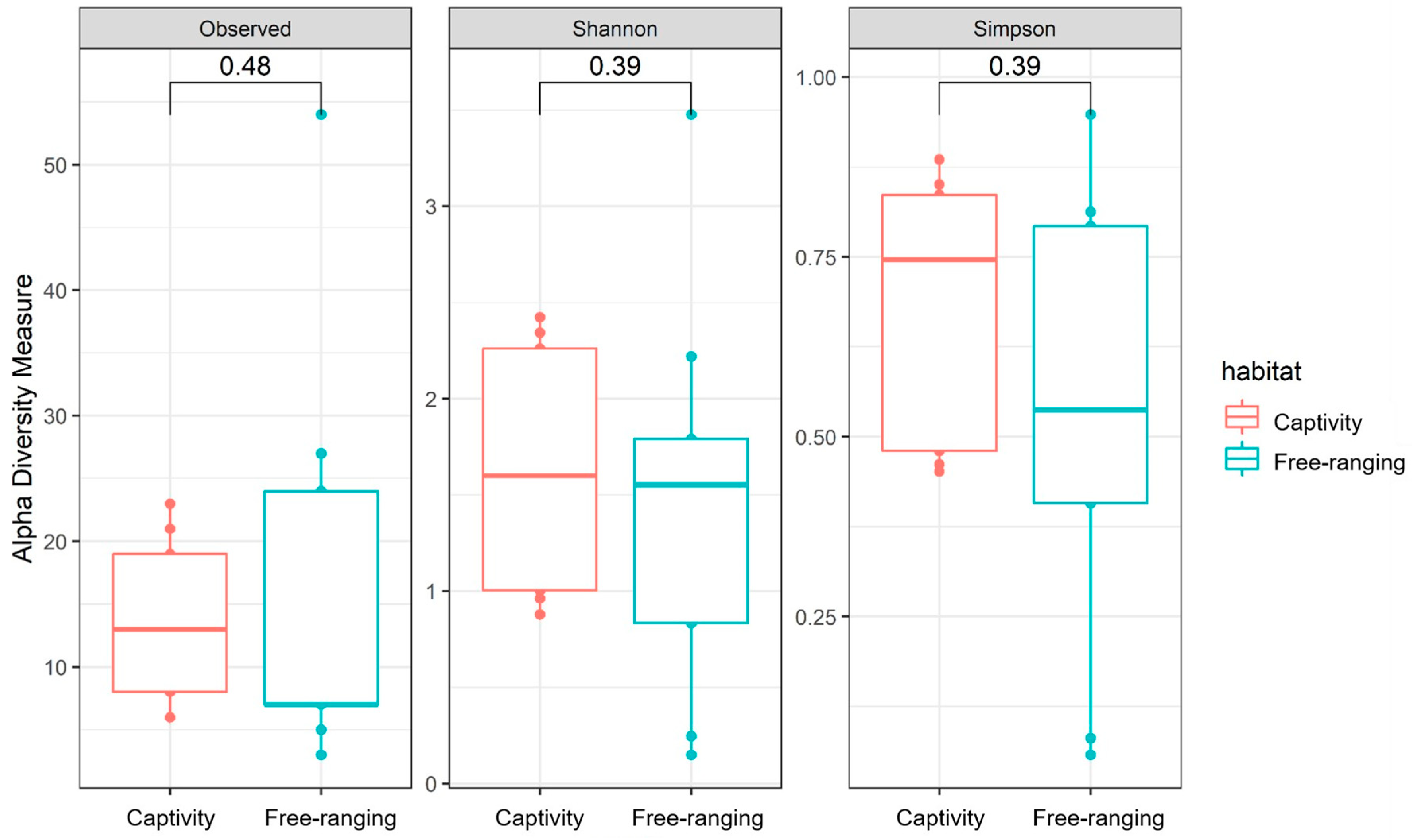
3.1. Diversity Metrics of the Fecal Samples’ Bacterial Communities
3.2. Taxonomic Profile, Relative Abundance, and Differential Abundance of Bacterial Community Present in the Feces from Captive and Wild Saltator Similis
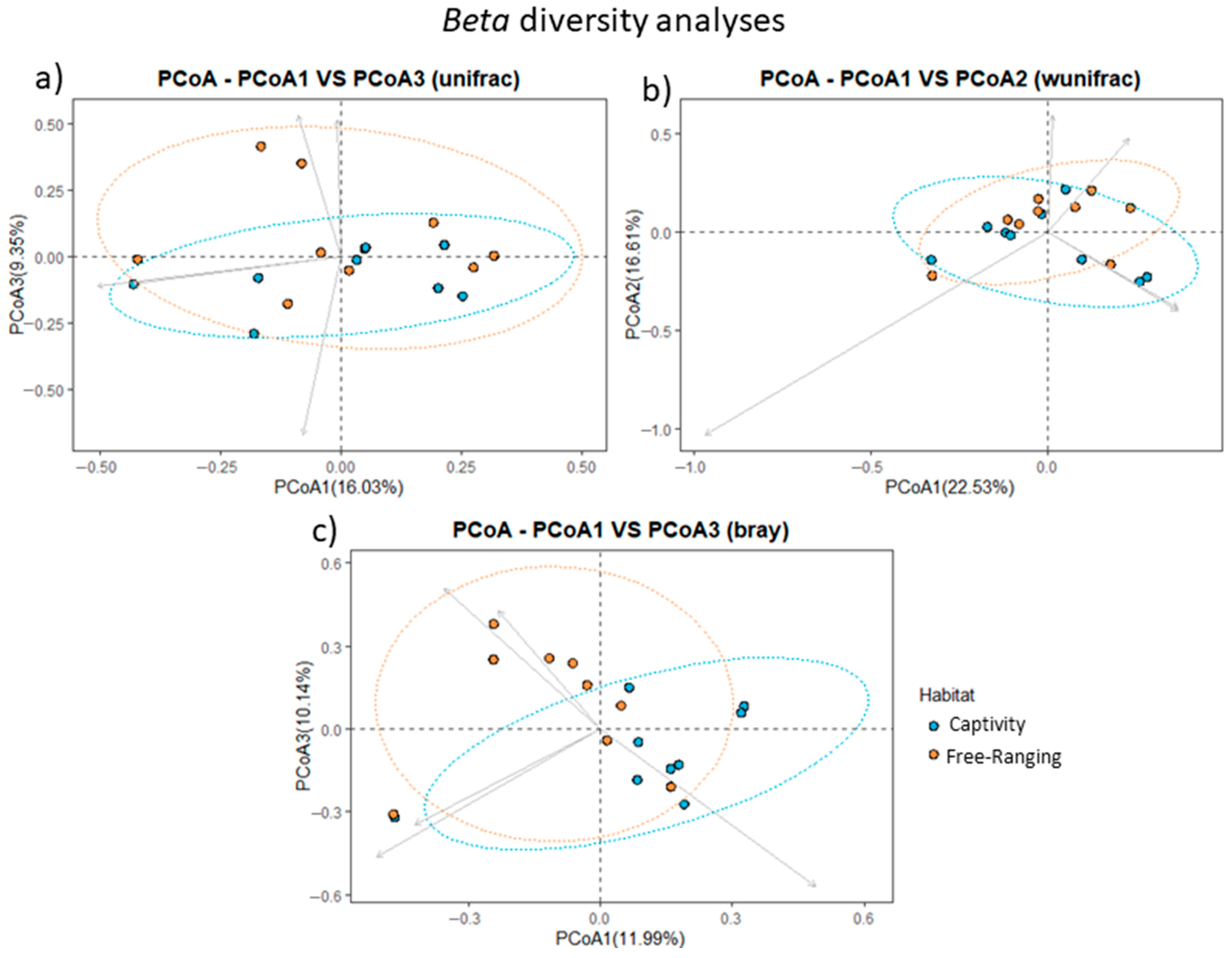
3.3. Fecal Bacterial Community Profile Is Guided by the Habitat
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Macke, E.; Tasiemski, A.; Massol, F.; Callens, M.; Decaestercker, E. Life history and eco-evolutionary dynamics in light of the gut microbiota. Oikos 2017, 126, 508–531. [Google Scholar] [CrossRef]
- Montoya-Ciriaco, N.; Gómez-Acata, S.; Muñoz-Arenas, L.C.; Dendooven, L.; Estrada-Torres, A.; De La Vega-Pérez, A.H.D.; Navarro-Noya, Y.E. Dietary effects on gut microbiota of the mesquite lizard Sceloporus grammicus (Wiegmann, 1828) across different altitudes. Microbiome 2020, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.M.; Nguyen, B.N.; Neumann, L.M.; Miller, M.; Buss, P.; Daniels, S.; Ahn, M.J.; Crandall, K.A.; Pukazhenthi, B. Gut microbiome diferences between wild and captive black rhinoceros—Implications for rhino health. Nature 2019, 9, 7570. [Google Scholar] [CrossRef]
- Hamaya, R.; Ono, Y.; Chida, Y.; Inokuchi, R.; Kikuchi, K.; Tameda, T.; Tase, C.; Shinohara, K. Polytetrafluoroethylene fume–induced pulmonary edema: A case report and review of the literature. J. Med. Case Rep. 2015, 9, 111. [Google Scholar] [CrossRef]
- Berlow, M.; Wada, H.; Derryberry, E.P. Experimental Exposure to Noise Alters Gut Microbiota in a Captive Songbird. Microb. Ecol. 2022, 84, 1264–1277. [Google Scholar] [CrossRef]
- Solomon, G.; Love, A.C.; Vaziri, G.J.; Harvey, J.; Verrett, T.; Chernicky, K.; Simons, S.; Albert, L.; Chaves, J.A.; Knutie, S.A. Effect of urbanization and parasitism on the gut microbiota of Darwin’s finch nestlings. Mol Ecol. 2023, 32, 6059–6069. [Google Scholar] [CrossRef]
- Cohen, S.; Gianaros, P.J.; Manuck, S.B. A stage model of stress and disease. Perspect. Psychol. Sci. 2016, 11, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 46, 1282–1287. [Google Scholar] [CrossRef]
- Lyra, R.B.; Monteiro, L.R.; Ruiz-Miranda, R.C. Song as a signal of male identity and quality in the Green-winged Saltator (Saltator similis). Wilson J. Ornithol. 2022, 134, 86–96. [Google Scholar] [CrossRef]
- Osbrink, A.; Meatte, M.A.; Tran, A.; Katri, K.; Herranen, K.K.; Meek, L.; Murakami-Smith, M.; Ito, J.; Bhadra, S.; Nunnenkamp, C.; et al. Traffic noise inhibits cognitive performance in a songbird. Proc. R. Soc. B 2021, 288, 20202851. [Google Scholar] [CrossRef]
- Cruz, C.E.F.; Soares, C.E.S.; Hirt, G.B.; Wagner, P.G.C.; Andretta, I.; Neto, W.N.C. Wild animals housed in the IBAMA Triage Center in Southern Brazil, 2005–2021, A glimpse into the endless conflicts between man and other animals. Ethnobiol. Conserv. 2022, 11, 29. [Google Scholar] [CrossRef]
- Cruz, C.E.F.; Wagner, P.G.C.; Driemeier, D.; Andretta, I. Live decoys: An old but effective tool for attracting, capturing, and studying free-living passerines. Eur. J. Wildl. 2022, 68, 24. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2, High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Gavin, A.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- McKnight, D.T.; Huerlimann, R.; Bower, D.S.; Schwarzkopf, L.; Alford, R.A.; Zenger, K.R. microDecon: A highly accurate read-subtraction tool for the post-sequencing removal of contamination in metabarcoding studies. Environ. DNA 2019, 1, 14–25. [Google Scholar] [CrossRef]
- Lahti, L.; Shetty, S.; Taruga, N.; Leung, E.; Gilmore, R.; Salöjarvi, J.; Blake, T.; Obenchain, V.; Pagès, H.; Ramos, M. Tools for Microbiome Analysis in R. (Version 2.1.28). Bioc Package. 2017. Available online: http://microbiome.github.io/microbiome (accessed on 25 October 2023).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Wilcoxon, F. Some Uses of Statistics in Plant Pathology. IBS 1945, 1, 41–45. [Google Scholar] [CrossRef]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Simpson, G.L.; Solymos, P.; Henry, M.H.; Stevens, H.W. Community Ecology Package—The Vegan Package (Version 1.15-1). BCI. 2008. Available online: http://vegan.r-forge.r-project.org/ (accessed on 25 October 2023).
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Sottas, C.; Schmiedova, L.; Kreisinger, J.; Albrecht, T.; Reif, J.; Osiejuk, T.S.; Reifova, R. Gut microbiota in two recently diverged passerine species: Evaluating the effects of species identity, habitat use and geographic distance. BMC Ecol. Evol. 2021, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Hedblom, G.A.; Dev, K.; Bowden, S.D. Draft genome sequence of “Candidatus Arthromitus” UMNCA01, a suspected commensal isolated from the gut microbiome of commercial turkey. ASM 2020, 9, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shao, H.; Peng, X.; Liu, T.; Tan, Z. Microbial characteristics colonized in intestinal mucosa of mice with diarrhea and repeated stress. 3 Biotech 2020, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Del-Pozo, J.; Turnbull, J.; Ferguson, H.; Crumlish, M. A comparative molecular study of the presence of ‘‘Candidatus arthromitus’’ in the digestive system of rainbow trout, Oncorhynchus mykiss (Walbaum), healthy and affected with rainbow trout gastroenteritis. J. Fish Dis. 2010, 33, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sancho, M.; Cerdá, I.; Fernández-Bravo, A.; Domínguez, L.; Figueras, M.J.; Fernández-Garayzábal, J.F.; Vela, A.I. Limited performance of MALDI-TOF for identification of fish Aeromonas isolates at species level. J. Fish Dis. 2018, 10, 1485–1493. [Google Scholar] [CrossRef]
- Sharma, D.; Patel, A.; Soni, P.; Sharma, P.; Gupta, B. Empedobacter brevis meningitis in a neonate: A very rare case of neonatal meningitis and literature review. Pediatr Surg Case Rep. 2016, 2016, 7609602. [Google Scholar] [CrossRef]
- Kim, Y.O.; Park, S.; Park, I.S.; Nam, B.H.; Kim, D.G.; Yoon, J.H. Empedobacter tilapiae sp. nov., isolated from an intestine of Nile tilapia (Oreochromis niloticus). Int. J. Syst. Evol. Microbiol. 2019, 69, 2781–2786. [Google Scholar] [CrossRef]
- Falagán, C.; Johnson, D.B. Acidibacter ferrireducens gen. nov., sp. nov.: An acidophilic ferric iron-reducing gammaproteobacterium. Extremophiles 2014, 18, 1067–1073. [Google Scholar] [CrossRef]
- Wan, K.; Gou, L.; Ye, C.; Zhu, J.; Zhang, M.; Yu, X. Accumulation of antibiotic resistance genes in full-scale drinking water biological activated carbon (BAC) filters during backwash cycles. Water Res. 2021, 190, 116744. [Google Scholar] [CrossRef]
- Wang, D.; Wei, C. Bacterial communities in digestive and excretory organs of cicadas. Arch. Microbiol. 2020, 202, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Perez, S.; Baker, L.J.; Morris, M.M.; Tsuji, K.; Sanchez, V.A.; Fukami, T.; Vannette, R.L.B.; Hendry, T.A. Acinetobacter pollinis sp. nov., Acinetobacter baretiae sp. nov. and Acinetobacter rathckeae sp. nov., isolated from floral nectar and honey bees. Int. J. Syst. Evol. Microbiol. 2021, 71, 4783. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Wang, H.; Yu, Z.; Zhang, S.; Qi, X.; Sun, L.; Wang, Z.; Zhang, M.; Ahmed, T.; Li, B. Effect of two kinds of fertilizers on growth and rhizosphere soil properties of bayberry with decline disease. Plants 2021, 10, 2386. [Google Scholar] [CrossRef]
- Dey, D.K.; Park, J.; Kang, S.C. Genotypic, phenotypic, and pathogenic characterization of the soil isolated Acinetobacter courvalinii. Microb. Pathog. 2020, 149, 104287. [Google Scholar] [CrossRef] [PubMed]
- Paganin, P.; Alisi, C.; Dore, E.; Fancello, D.; Marras, P.A.; Medas, D.; Montereali, M.R.; Naitza, S.; Rigonat, N.; Sprocati, A.R.; et al. Microbial diversity of bacteria involved in biomineralization processes in mine-impacted freshwaters. Front. Microbiol. 2021, 12, 778199. [Google Scholar] [CrossRef]
- Chen, W.M.; You, Y.X.; Young, C.C.; Lin, S.Y.; Sheu, S.Y. Flavobacterium difficile sp. nov., isolated from a freshwater waterfall. Arch. Microbiol. 2021, 203, 4449–4459. [Google Scholar] [CrossRef]
- Le, V.V.; Lee, H.; Padakandla, S.R.; Cha, I.T.; Lee, K.E.; Chae, J.C. Flavobacterium inviolabile sp. nov. isolated from stream water. Arch. Microbiol. 2021, 203, 3633–3639. [Google Scholar] [CrossRef]
- Liu, B.; Yang, X.; Sheng, M.; Yang, Z.; Qiu, J.; Wang, C.; He, J. Sphingobacterium olei sp. nov., isolated from oil-contaminated soil. Int. J. Syst. Evol. Microbiol. 2020, 70, 1931–1939. [Google Scholar] [CrossRef]
- Song, J.; Joung, Y.; Li, S.H.; Hwang, J.; Cho, J.C. Sphingobacterium chungjuense sp. nov., isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 2020, 70, 6126–6132. [Google Scholar] [CrossRef]
- Góngora, E.; Elliott, K.H.; Whyte, L. Gut microbiome is affected by inter-sexual and inter-seasonal variation in diet for thick-billed murres (Uria lomvia). Nature 2021, 11, 1200. [Google Scholar] [CrossRef]
- Braun, M.S.; Wang, E.; Zimmermann, S.; Wagner, H.; Wink, M. Kocuria tytonis sp. nov., isolated from the uropygial gland of an American barn owl (Tyto furcata). Int. J. Syst. Evol. Microbiol. 2019, 69, 447–451. [Google Scholar] [CrossRef]
- Brown, K.I.; Boehm, A.B. Comparative decay of Catellicoccus marimmalium and enterococci in beach sand and seawater. Water Res. 2015, 83, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Marion, J.W.; Lee, J. Development and application of a quantitative PCR assay targeting Catellicoccus marimammalium for assessing gull-associated fecal contamination at Lake Erie beaches. Sci. Total Environ. 2013, 8, 454–455. [Google Scholar] [CrossRef]
- Zambon, J.J. Actinobacillus actinomycetemcomitans in human periodontal disease. J. Clin. Periodontol. 1985, 12, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, M.; Hunt, B.; Carson, T.; Duggett, N.; Whatmore, A.M. Actinobacillus vicugnae sp. nov., isolated from alpaca (Vicugna pacos). Int. J. Syst. Evol. Microbiol. 2019, 69, 3170–3177. [Google Scholar] [CrossRef]
- Bisgaard, M.; Christensen, H. Classification of avian haemolytic Actinobacillus-like organisms (Bisgaard taxon 26) associated with anseriforme birds as Actinobacillus anseriformium sp. nov. Int. J. Syst. Evol. Microbiol. 2012, 62, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Tonouchi, A.; Kitamura, K.; Fujita, T. Brevibacterium yomogidense sp. nov., isolated from a soil conditioner made from poultry manure. Int. J. Syst. Evol. Microbiol. 2013, 63, 516–520. [Google Scholar] [CrossRef]
- Deng, T.; Lu, H.; Qian, Y.; Chen, X.; Yang, X.; Guo, J.; Sun, G.; Xu, M. Brevibacterium rongguiense sp. nov., isolated from freshwater sediment. Int. J. Syst. Evol. Microbiol. 2020, 70, 5205–5210. [Google Scholar] [CrossRef]
- Pei, S.; Niu, S.; Xie, F.; Wang, W.; Zhang, S.; Zhang, G. Brevibacterium limosum sp. nov., Brevibacterium pigmenatum sp. nov., and Brevibacterium atlanticum sp. nov., three novel dye decolorizing actinobacteria isolated from ocean sediments. J. Microbiol. 2021, 59, 898–910. [Google Scholar] [CrossRef]
- Gomard, Y.; Flores, O.; Vittecoq, M.; Blanchon, T.; Toty, C.; Duron, O.; Mavingui, P.; Tortosa, P.; McCoy, K. Changes in Bacterial diversity, composition and interactions during the development of the seabird tick Ornithodoros maritimus (Argasidae). Microb. Ecol. 2021, 81, 770–783. [Google Scholar] [CrossRef]
- Mohan, K. Brevibacterium sp. from poultry. Anton. Leeuw. Int. J. 1981, 47, 449–453. [Google Scholar] [CrossRef]
- Pascual, C.; Collins, M.D. Brevibacterium aviurn sp. nov., isolated from poultry. Int. J. Syst. Bacteriol. 1999, 49, 1527–1530. [Google Scholar] [CrossRef]
- Zhou, Q.; Lan, F.; Li, X.; Yan, W.; Sun, C.; Li, J.; Yang, N.; Wen, C. The spatial and temporal characterization of gut microbiota in broilers. Front. Vet. Sci. 2021, 8, 712226. [Google Scholar] [CrossRef]
- Aguilar-Lopez, M.; Wetzel, C.; MacDonald, A.; Ho, T.T.B.; Donovan, S.M. Human milk-based or bovine milk-based fortifiers differentially impact the development of the gut microbiota of preterm infants. Front. Pediatr. 2021, 9, 719096. [Google Scholar] [CrossRef]
- Joyner, J.; Wanless, D.; Sinigalliano, C.D.; Lipp, E.K. Use of Quantitative Real-Time PCR for direct detection of Serratia marcescens in marine and other aquatic environments. Appl. Environ. Microbiol. 2014, 80, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Castillo, D.; Power, P.; Cerdeira, L.; Cardenas-Arias, A.; Moura, Q.; Oliveira, F.A.; Levy, C.E.; Gutkind, G.; Catão-Dias, J.L.; Lincopan, N. FONA-7, a novel Extended-Spectrum b-Lactamase variant of the FONA family identified in Serratia fonticola. Microb. Drug Resist. 2020, 27, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Ruden, R.M.; Adelman, J.S. Disease tolerance alters host competence in a wild songbird. Biol. Lett. 2021, 17, 20210362. [Google Scholar] [CrossRef] [PubMed]
- Mohan, T.; Farid, N.S.S.; Swathi, K.V.; Sowmya, A.; Ramani, K. Sustainable biological system for the removal of high strength ammoniacal nitrogen and organic pollutants in poultry waste processing industrial effluent. J. AWMA 2020, 70, 1236–1243. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Code | Origin (City) | Habitat | General Health Status | Life Stage | Raw Reads | Filtered Reads | Non-Chimeric Reads (%) |
|---|---|---|---|---|---|---|---|
| 1 | Barra do Ribeiro | Wild | Healthy | Adult | 275,096 | 260,926 | 73.75 |
| 2 | Barra do Ribeiro | Wild | Healthy | Adult | 202,753 | 193,485 | 79.92 |
| 3 | Barra do Ribeiro | Wild | Healthy | Adult | 195,656 | 187,601 | 92.33 |
| 4 | Barra do Ribeiro | Wild | Healthy | Adult | 248,750 | 236,447 | 91.41 |
| 5 | Barra do Ribeiro | Wild | Healthy | Adult | 46,064 | 43,080 | 90.69 |
| 6 | Eldorado do Sul | Wild | Healthy | Adult | 184,185 | 175,746 | 50.38 |
| 7 | Eldorado do Sul | Wild | Healthy | Adult | 3713 | 2995 | 59.14 |
| 8 | Eldorado do Sul | Wild | Healthy | Adult | 253,852 | 243,964 | 80.79 |
| 9 | Eldorado do Sul | Wild | Healthy | Adult | 237,229 | 230,719 | 84.64 |
| 10 | Porto Alegre | Captivity | Healthy | Adult | 224,010 | 217,867 | 85.80 |
| 11 | Porto Alegre | Captivity | Healthy | Adult | 198,628 | 193,330 | 94.00 |
| 12 | Porto Alegre | Captivity | Healthy | Adult | 189,314 | 181,439 | 85.92 |
| 13 | Porto Alegre | Captivity | Healthy | Adult | 152,275 | 146,937 | 76.68 |
| 14 | Porto Alegre | Captivity | Healthy | Adult | 207,188 | 193,388 | 67.68 |
| 15 | Porto Alegre | Captivity | Healthy | Adult | 260,863 | 246,147 | 80.81 |
| 16 | Porto Alegre | Captivity | Healthy | Adult | 162,525 | 157,993 | 70.63 |
| 17 | Porto Alegre | Captivity | Healthy | Adult | 210,880 | 201,375 | 75.81 |
| 18 | Porto Alegre | Captivity | Healthy | Adult | 224,236 | 216,852 | 74.28 |
| Markers | Genus | Habitat | Effect Linear Discriminant Analysis | p Value | p Adjusted |
|---|---|---|---|---|---|
| Marker 1 | “Candidatus Arthromitus” | Captivity | 5,337,960 | 0.046972020 | 0.046972020 |
| Marker 2 | Acinetobacter | Captivity | 5,236,298 | 0.001221026 | 0.001221026 |
| Marker 3 | Kocuria | Captivity | 4,664,164 | 0.023832113 | 0.023832113 |
| Marker 4 | Paracoccus | Captivity | 3,428,577 | 0.011922794 | 0.011922794 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zitelli, L.C.; Breyer, G.M.; Torres, M.C.; Menetrier, L.d.C.; Varela, A.P.M.; Mayer, F.Q.; Cruz, C.E.F.; Siqueira, F.M. Comparative Analysis of How the Fecal Microbiota of Green-Winged Saltator (Saltator similis) Diverge among Animals Living in Captivity and in Wild Habitats. Animals 2024, 14, 937. https://doi.org/10.3390/ani14060937
Zitelli LC, Breyer GM, Torres MC, Menetrier LdC, Varela APM, Mayer FQ, Cruz CEF, Siqueira FM. Comparative Analysis of How the Fecal Microbiota of Green-Winged Saltator (Saltator similis) Diverge among Animals Living in Captivity and in Wild Habitats. Animals. 2024; 14(6):937. https://doi.org/10.3390/ani14060937
Chicago/Turabian StyleZitelli, Larissa Caló, Gabriela Merker Breyer, Mariana Costa Torres, Luiza de Campos Menetrier, Ana Paula Muterle Varela, Fabiana Quoos Mayer, Cláudio Estêvão Farias Cruz, and Franciele Maboni Siqueira. 2024. "Comparative Analysis of How the Fecal Microbiota of Green-Winged Saltator (Saltator similis) Diverge among Animals Living in Captivity and in Wild Habitats" Animals 14, no. 6: 937. https://doi.org/10.3390/ani14060937
APA StyleZitelli, L. C., Breyer, G. M., Torres, M. C., Menetrier, L. d. C., Varela, A. P. M., Mayer, F. Q., Cruz, C. E. F., & Siqueira, F. M. (2024). Comparative Analysis of How the Fecal Microbiota of Green-Winged Saltator (Saltator similis) Diverge among Animals Living in Captivity and in Wild Habitats. Animals, 14(6), 937. https://doi.org/10.3390/ani14060937