
Comparative Analysis of Gut Microbiota Diversity Across Different Digestive Tract Sites in Ningxiang Pigs

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Animals and Gut Tissues Storage
2.3. 16S rRNA Library Construction
2.4. 16S rRNA Sequencing Data Preprocessing
2.5. Species Annotation Methods
2.6. Alpha Diversity Analysis and Beta Diversity Analysis
2.7. Significance Analysis of Intergroup Differences
2.8. PICRUSt2 Function Prediction
2.9. Statistical Analysis
3. Results
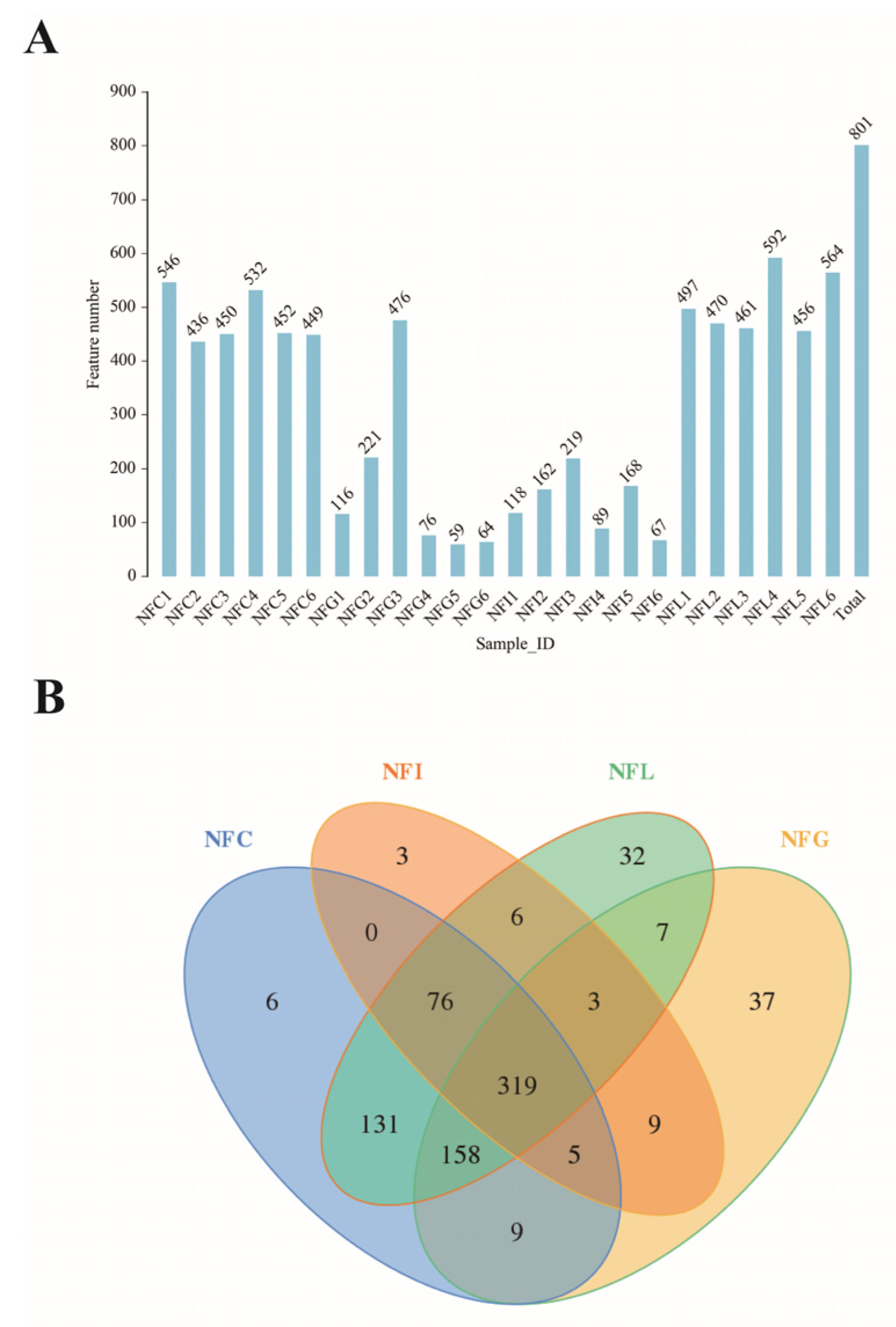
3.1. Microbial Community Composition and Diversity Across Nutrient Absorption Tissues
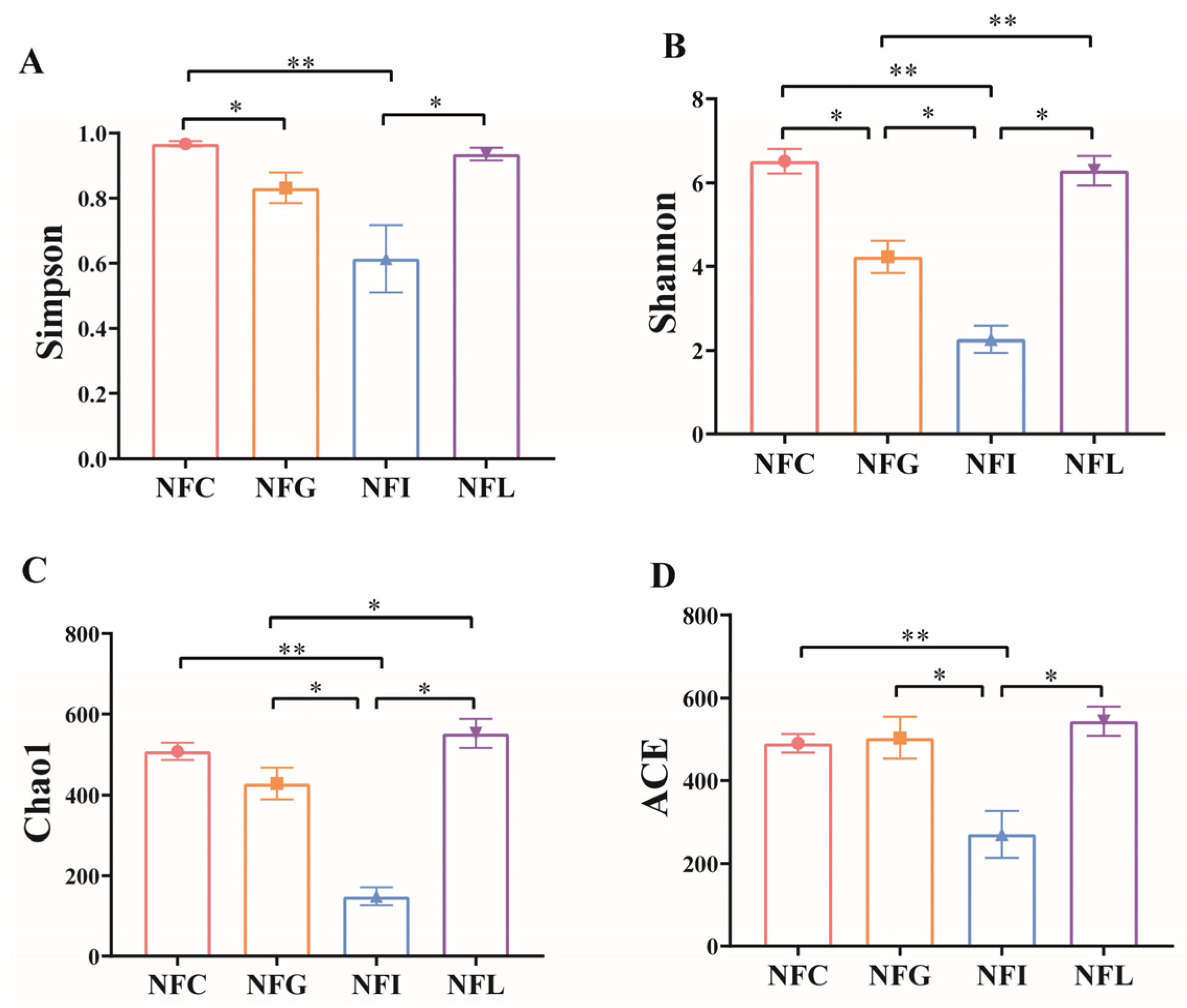
3.2. Sequencing Saturation and Alpha Diversity of Microbial Profiles
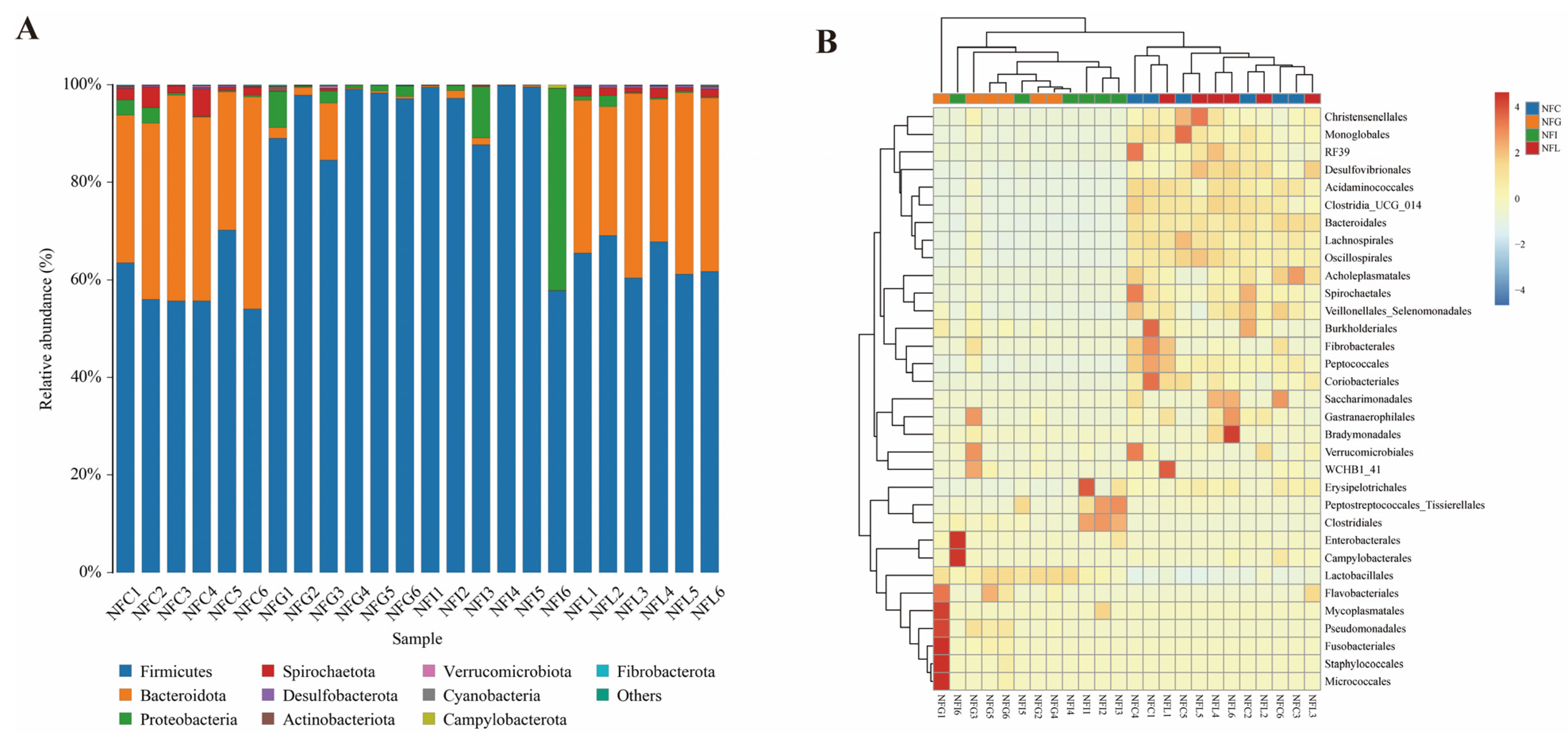
3.3. Microbial Composition and Abundance Profiles in NFC and NFL Reveal Functional Similarities
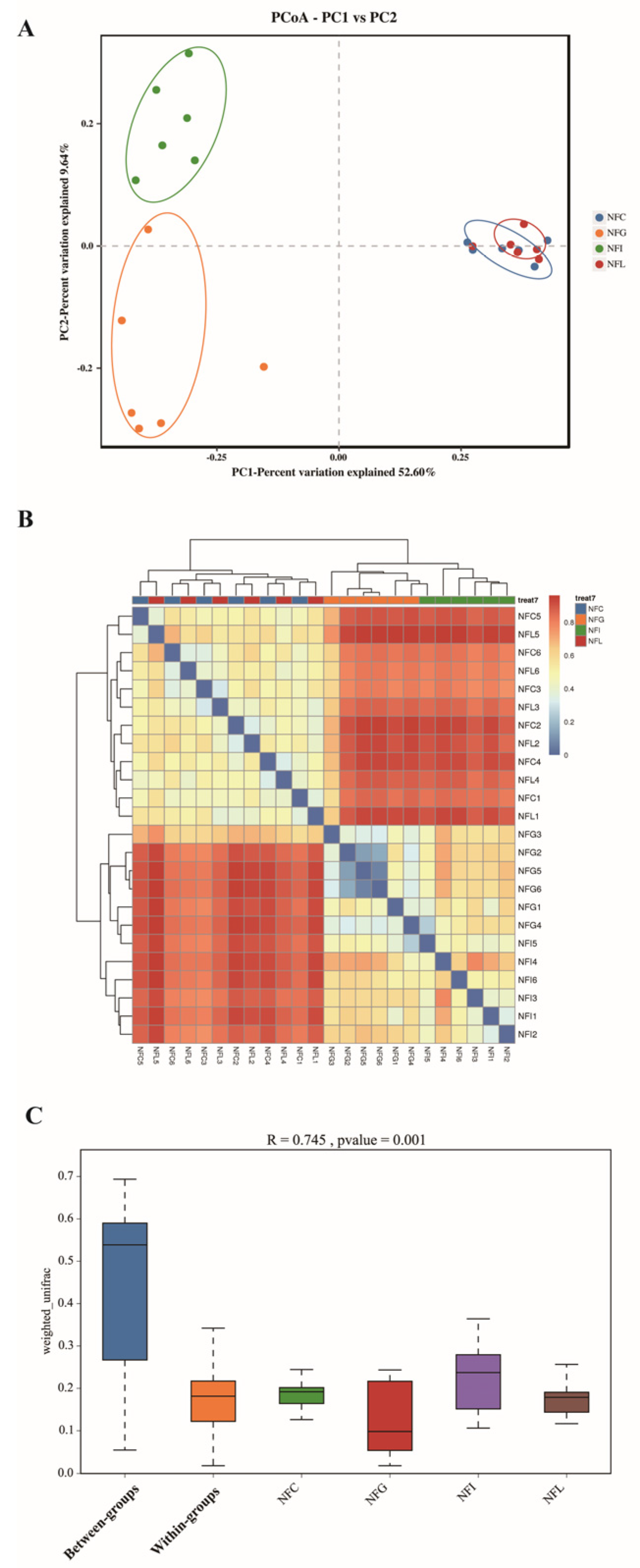
3.4. Beta Diversity of Microbial Profiles
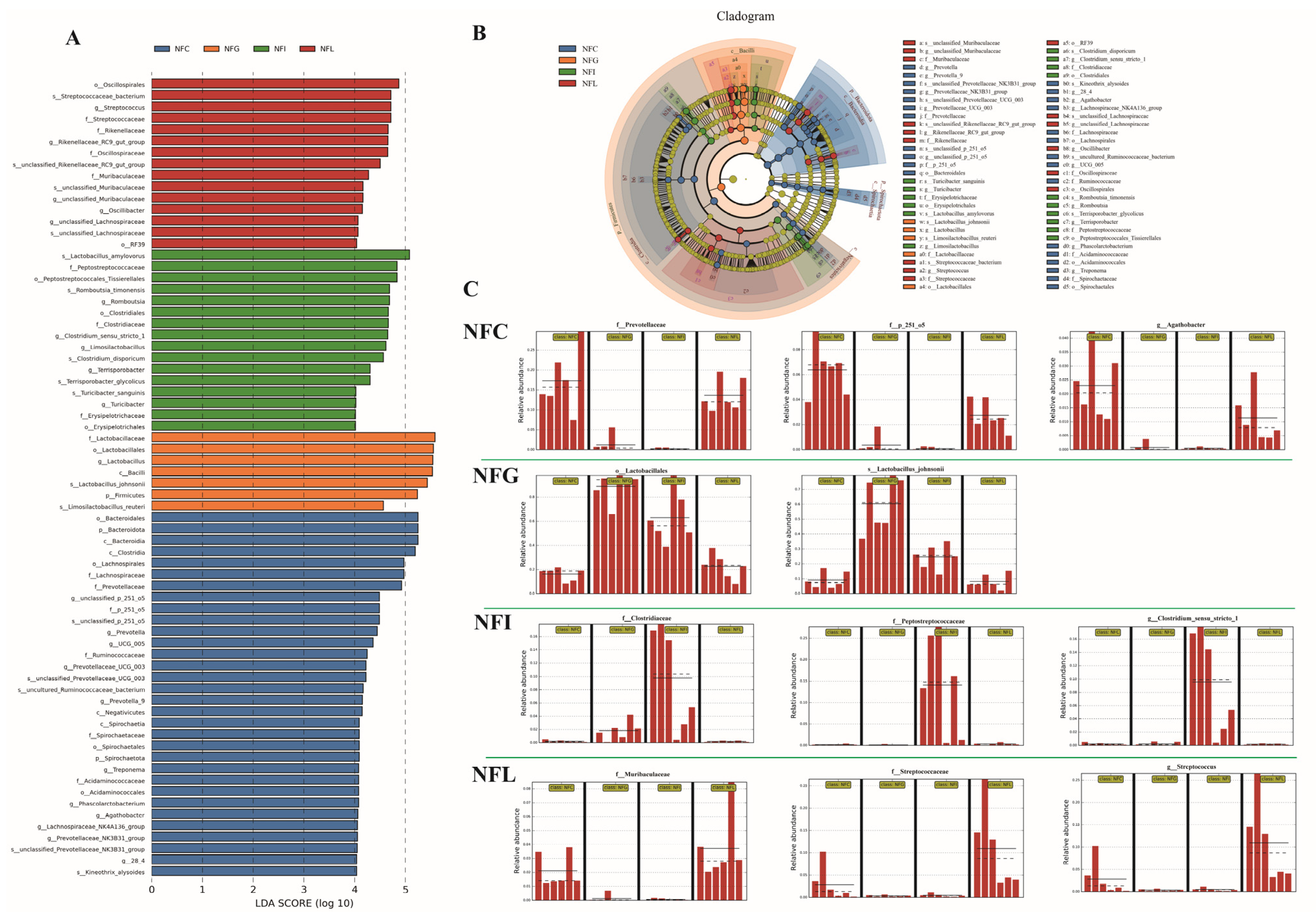
3.5. LEfSe Analysis Reveals Key Microbial Biomarkers in Nutrient Absorption Tissues
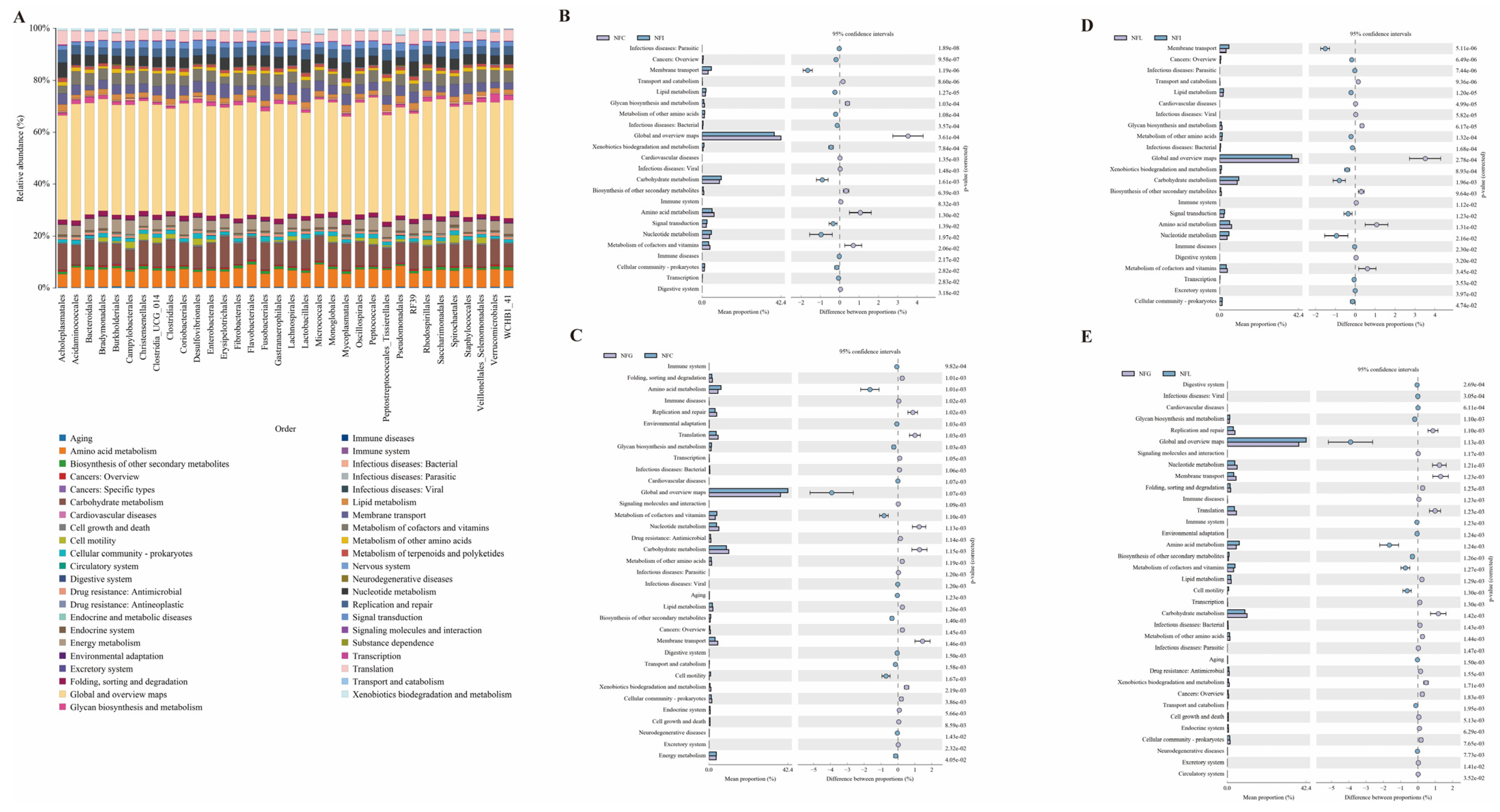
3.6. Functional Profiling and Comparative Metabolic Analysis of Microbial Communities Across Nutrient Absorption Tissues
4. Discussion
4.1. Analysis of α and β Diversity of Microbial Communities
4.2. Comparative Analysis of Microbial Community Composition Across Nutrient Tissues
4.3. Characterization of Tissue-Specific Microbes and Differential Metabolic Pathways in Nutrient Tissues
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drewnowski, A. Perspective: The place of pork meat in sustainable healthy diets. Adv. Nutr. 2024, 15, 100213. [Google Scholar] [CrossRef] [PubMed]
- Donohue, M.; Cunningham, D.L. Effects of grain and oilseed prices on the costs of US poultry production. J. Appl. Poult. Res. 2009, 18, 325–337. [Google Scholar] [CrossRef]
- Kellner, T.A.; Pilcher, C.M. 20 How do Producers and Nutritionists Determine Which Levers to Pull to Optimize Feed Cost and Profitability Under Differing Economic Conditions? J. Anim. Sci. 2023, 101 (Suppl. S2), 17–18. [Google Scholar] [CrossRef]
- Lei, L.; Wang, Z.; Li, J.; Yang, H.; Yin, Y.; Tan, B.; Chen, J. Comparative microbial profiles of colonic digesta between Ningxiang pig and large white pig. Animals 2021, 11, 1862. [Google Scholar] [CrossRef]
- Li, H.; Ma, L.; Li, Z.; Yin, J.; Tan, B.; Chen, J.; Ma, X. Evolution of the gut microbiota and its fermentation characteristics of Ningxiang pigs at the young stage. Animals 2021, 11, 638. [Google Scholar] [CrossRef]
- Wang, T.Y.; Tao, S.Y.; Wu, Y.X.; An, T.; Lv, B.H.; Liu, J.X.; Jiang, G.J. Quinoa reduces high-fat diet-induced obesity in mice via potential microbiota-gut-brain-liver interaction mechanisms. Microbiol. Spectr. 2022, 10, e00329-22. [Google Scholar] [CrossRef] [PubMed]
- Pannaraj, P.S.; Li, F.; Cerini, C.; Bender, J.M.; Yang, S.; Rollie, A.; Aldrovandi, G.M. Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr. 2017, 171, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Schloss, P.D. Dynamics and associations of microbial community types across the human body. Nature 2014, 509, 357–360. [Google Scholar] [CrossRef]
- Betancur-Murillo, C.L.; Aguilar-Marín, S.B.; Jovel, J. Prevotella: A key player in ruminal metabolism. Microorganisms 2022, 11, 1. [Google Scholar] [CrossRef]
- Zou, P.; Yang, F.; Ding, Y.; Zhang, D.; Liu, Y.; Zhang, J.; Wang, Y. Lipopolysaccharide downregulates the expression of ZO-1 protein through the Akt pathway. BMC Infect. Dis. 2022, 22, 774. [Google Scholar] [CrossRef]
- Utepbergenov, D.I.; Fanning, A.S.; Anderson, J.M. Dimerization of the scaffolding protein ZO-1 through the second PDZ domain. J. Biol. Chem. 2006, 281, 24671–24677. [Google Scholar] [PubMed]
- Kuo, W.T.; Odenwald, M.A.; Turner, J.R.; Zuo, L. Tight junction proteins occludin and ZO-1 as regulators of epithelial proliferation and survival. Ann. N. Y. Acad. Sci. 2022, 1514, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.B.; McGinn, L.S.; Chen, Z. Gut microbiota amplifies host-intrinsic conversion from the CD8 T cell lineage to CD4 T cells for induction of mucosal immune tolerance. Gut Microbes 2016, 7, 40–47. [Google Scholar] [CrossRef]
- Lakho, S.A.; Haseeb, M.; Huang, J.; Yang, Z.; Hasan, M.W.; Aleem, M.T.; Li, X. Glyceraldehyde-3-phosphate dehydrogenase from Eimeria acervulina modulates the functions of chicken dendritic cells to boost Th1 type immune response and stimulates autologous CD4+ T cells differentiation in-vitro. Vet. Res. 2020, 51, 138. [Google Scholar] [PubMed]
- Wang, Q.; Sun, Q.; Qi, R.; Wang, J.; Qiu, X.; Liu, Z.; Huang, J. Effects of Lactobacillus plantarum on the intestinal morphology, intestinal barrier function and microbiota composition of suckling piglets. J. Anim. Physiol. Anim. Nutr. 2019, 103, 1908–1918. [Google Scholar]
- Hu, L.; Geng, S.; Li, Y.; Cheng, S.; Fu, X.; Yue, X.; Han, X. Exogenous fecal microbiota transplantation from local adult pigs to crossbred newborn piglets. Front. Microbiol. 2018, 8, 2663. [Google Scholar]
- Le Bon, M.; Tötemeyer, S.; Emes, R.D.; Mellits, K.H. Gut transcriptome reveals differential gene expression and enriched pathways linked to immune activation in response to weaning in pigs. Front. Genet. 2022, 13, 961474. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar]
- Zhou, Y.; Liu, Y.X.; Li, X. USEARCH 12: Open-source software for sequencing analysis in bioinformatics and microbiome. Imeta 2024, 3, e236. [Google Scholar]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [PubMed]
- Mohsen, A.; Park, J.; Chen, Y.A.; Kawashima, H.; Mizuguchi, K. Impact of quality trimming on the efficiency of reads joining and diversity analysis of Illumina paired-end reads in the context of QIIME1 and QIIME2 microbiome analysis frameworks. BMC Bioinform. 2019, 20, 581. [Google Scholar]
- Analysis and Visualization of Streaming Media Platforms Based on the R Language—Take Netflix as an Example. Available online: https://drpress.org/ojs/index.php/EHSS/article/view/2766 (accessed on 1 February 2025). [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Langille, M.G. PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv 2019, 672295. [Google Scholar]
- Li, X.; Song, Y.; Ma, X.; Zhang, Y.; Liu, X.; Cheng, L.; Sun, Q. Lactobacillus plantarum and Lactobacillus fermentum alone or in combination regulate intestinal flora composition and systemic immunity to alleviate obesity syndrome in high-fat diet rat. Int. J. Food Sci. Technol. 2018, 53, 137–146. [Google Scholar]
- Cao, X.; Tang, L.; Zeng, Z.; Wang, B.; Zhou, Y.; Wang, Q.; Li, W. Effects of probiotics BaSC06 on intestinal digestion and absorption, antioxidant capacity, microbiota composition, and macrophage polarization in pigs for fattening. Front. Vet. Sci. 2020, 7, 570593. [Google Scholar]
- Allen, J.M.; Jaggers, R.M.; Solden, L.M.; Loman, B.R.; Davies, R.H.; Mackos, A.R.; Bailey, M.T. Dietary oligosaccharides attenuate stress-induced disruptions in immune reactivity and microbial B-vitamin metabolism. Front. Immunol. 2019, 10, 1774. [Google Scholar]
- Ke, S.; Fang, S.; He, M.; Huang, X.; Yang, H.; Yang, B.; Chen, C.; Huang, L. Age-based dynamic changes of phylogenetic composition and interaction networks of health pig gut microbiome feeding in a uniformed condition. BMC Vet. Res. 2019, 15, 172. [Google Scholar]
- Alcazar, M.; Escribano, J.; Ferré, N.; Closa-Monasterolo, R.; Selma-Royo, M.; Feliu, A.; Balcells, E. Gut microbiota is associated with metabolic health in children with obesity. Clin. Nutr. 2022, 41, 1680–1688. [Google Scholar]
- Zhang, B.; Liu, M.; Yue, Z.; Chen, X.; Li, C.; Liu, L.; Li, F. Combined omics analysis further unveils the specific role of butyrate in promoting growth in early-weaning animals. Int. J. Mol. Sci. 2023, 24, 1787. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Cole, C.G.; Coyne, M.J.; Lin, H.; Dylla, N.; Smith, R.C.; Comstock, L.E. Comprehensive analyses of a large human gut Bacteroidales culture collection reveal species and strain level diversity and evolution. Cell Host Microbe 2024, 32, 1853–1867. [Google Scholar]
- Xu, B.; Yan, Y.; Yin, B.; Zhang, L.; Qin, W.; Niu, Y.; Ma, L. Dietary glycyl-glutamine supplementation ameliorates intestinal integrity, inflammatory response, and oxidative status in association with the gut microbiota in LPS-challenged piglets. Food Funct. 2021, 12, 3539–3551. [Google Scholar] [PubMed]
- Ariyoshi, T.; Hagihara, M.; Takahashi, M.; Mikamo, H. Effect of Clostridium butyricum on gastrointestinal infections. Biomedicines 2022, 10, 483. [Google Scholar] [CrossRef] [PubMed]
- Araki, H.; Kuriyama, T.; Nakagawa, K.; Karasawa, T. The microbial synergy of Peptostreptococcus micros and Prevotella intermedia in a murine abscess model. Oral Microbiol. Immunol. 2004, 19, 177–181. [Google Scholar]
- Van Dalen, P.J.; van Deutekom-Mulder, E.C.; De Graaff, J.; Van Steenbergen, T.J.M. Pathogenicity of Peptostreptococcus micros morphotypes and Prevotella species in pure and mixed culture. J. Med. Microbiol. 1998, 47, 135–140. [Google Scholar] [PubMed]
- Conrads, G.; Soffner, J.; Pelz, K.; Mutters, R. Taxonomic update and clinical significance of species within the genus Peptostreptococcus. Clin. Infect. Dis. 1997, 25, S94–S97. [Google Scholar]
- Shen, X.H.; Guan, J.; Lu, D.P.; Hong, S.C.; Yu, L.; Chen, X. Peptostreptococcus Anaerobius enhances dextran sulfate sodium-induced colitis by promoting nf-κB-NLRP3-Dependent macrophage pyroptosis. Virulence 2024, 15, 2435391. [Google Scholar]
- Xie, C.; Zhu, X.; Xu, B.; Niu, Y.; Zhang, X.; Ma, L.; Yan, X. Integrated analysis of multi-tissues lipidome and gut microbiome reveals microbiota-induced shifts on lipid metabolism in pigs. Anim. Nutr. 2022, 10, 280–293. [Google Scholar]
- Wu, C.; Lyu, W.; Hong, Q.; Zhang, X.; Yang, H.; Xiao, Y. Gut microbiota influence lipid metabolism of skeletal muscle in pigs. Front. Nutr. 2021, 8, 675445. [Google Scholar]
- Mainardi, E.; Corino, C.; Rossi, R. The effect of vitamins on the immune systems of pigs. Animals 2024, 14, 2126. [Google Scholar] [CrossRef]
- Geervliet, M.; de Vries, H.; Jansen, C.A.; Rutten, V.P.; van Hees, H.; Wen, C.; Wells, J.M. Effects of Escherichia coli Nissle 1917 on the porcine gut microbiota, intestinal epithelium and immune system in early life. Front. Microbiol. 2022, 13, 842437. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Zhang, X.; Fan, B.; Li, Y.; Zhao, S.; Guo, W.; He, W.; Zhao, Y.; Ni, Y.; Liu, M.; et al. Evaluation of the transcriptional regulatory efficacy of transcription regulatory sequences of porcine epidemic diarrhea virus. Vet. Microbiol. 2022, 267, 109376. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Zeng, X.; Wang, L.; Yin, L.; Wang, Q.; Yang, H. Comparative Analysis of Gut Microbiota Diversity Across Different Digestive Tract Sites in Ningxiang Pigs. Animals 2025, 15, 936. https://doi.org/10.3390/ani15070936
Li W, Zeng X, Wang L, Yin L, Wang Q, Yang H. Comparative Analysis of Gut Microbiota Diversity Across Different Digestive Tract Sites in Ningxiang Pigs. Animals. 2025; 15(7):936. https://doi.org/10.3390/ani15070936
Chicago/Turabian StyleLi, Wangchang, Xianglin Zeng, Lu Wang, Lanmei Yin, Qiye Wang, and Huansheng Yang. 2025. "Comparative Analysis of Gut Microbiota Diversity Across Different Digestive Tract Sites in Ningxiang Pigs" Animals 15, no. 7: 936. https://doi.org/10.3390/ani15070936
APA StyleLi, W., Zeng, X., Wang, L., Yin, L., Wang, Q., & Yang, H. (2025). Comparative Analysis of Gut Microbiota Diversity Across Different Digestive Tract Sites in Ningxiang Pigs. Animals, 15(7), 936. https://doi.org/10.3390/ani15070936