
Genetic Architecture of Hock Joint Bumps in Pigs: Insights from ROH and GWAS Analyses
 , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Genotyping and Quality Control
2.3. Homozygosity Analysis (ROH)
2.4. Heritability
2.5. GWAS
2.6. Functional Analysis
3. Results and Discussion
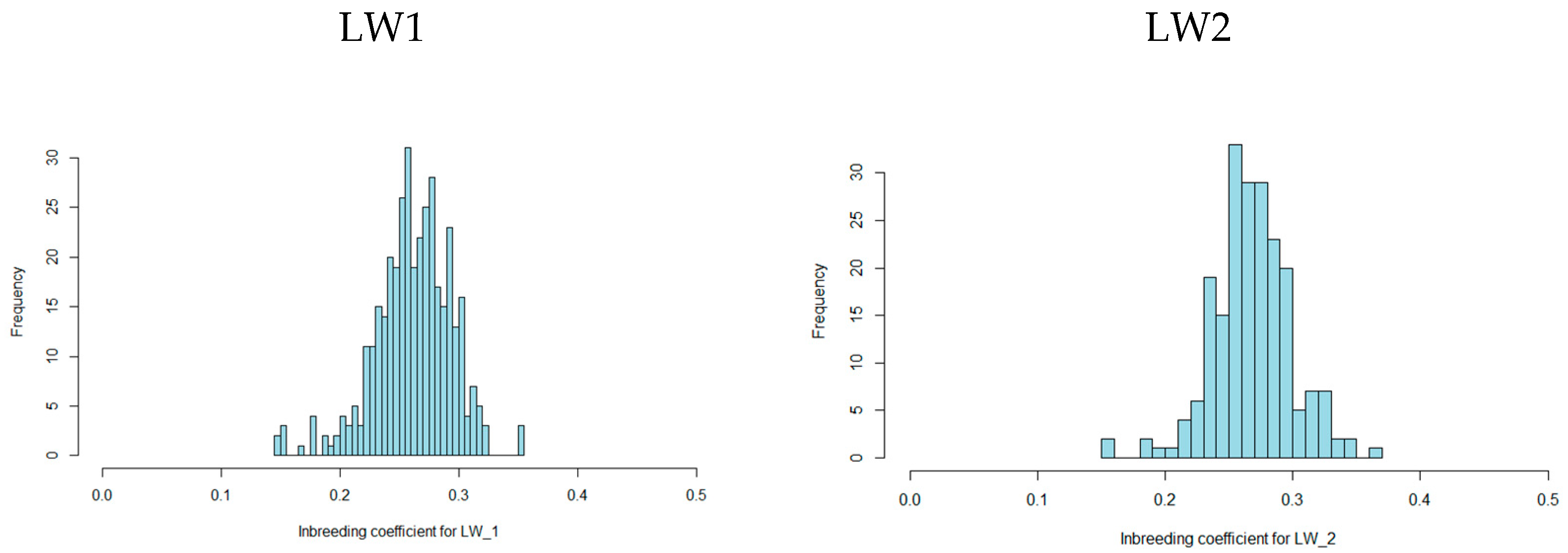
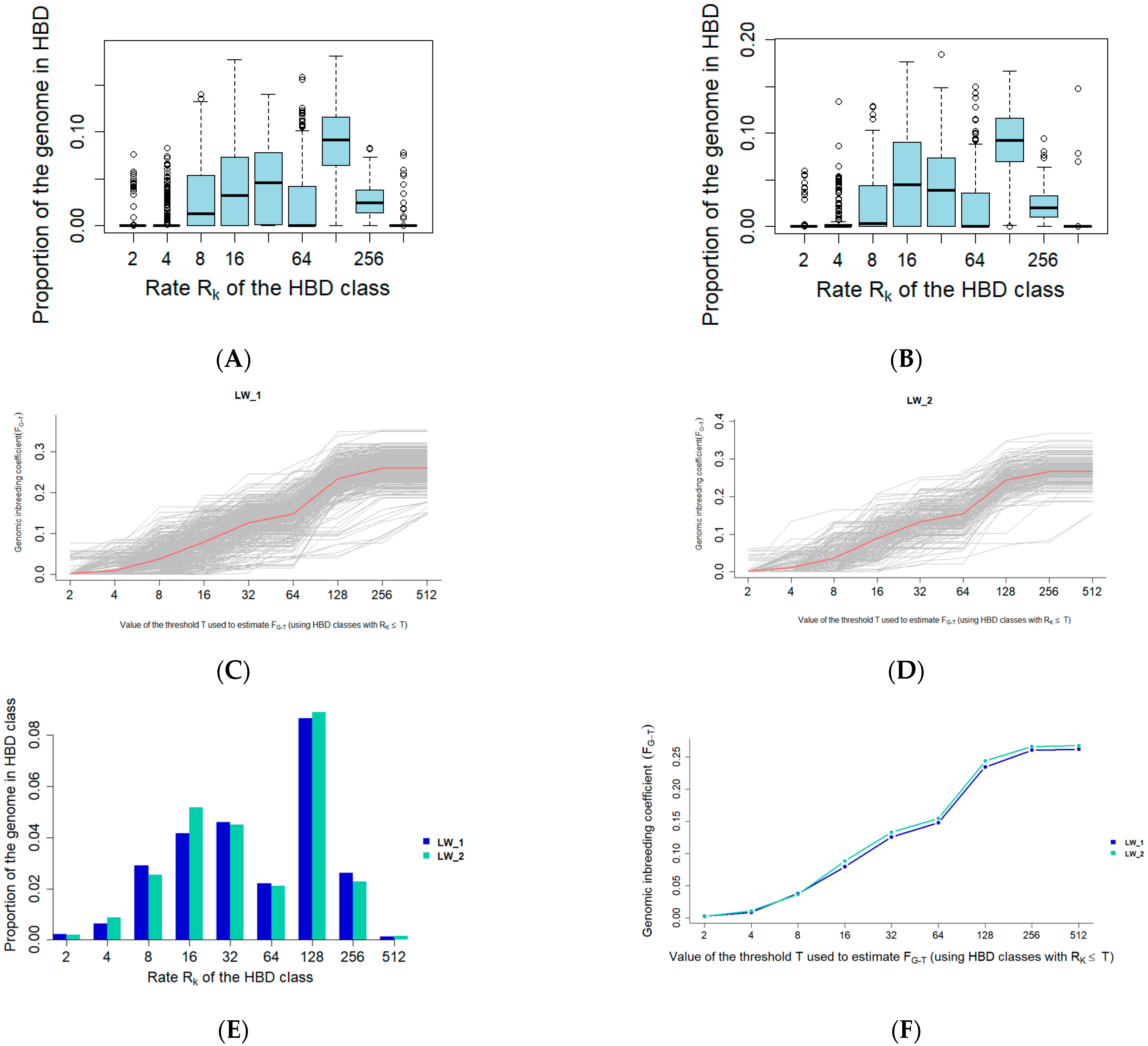
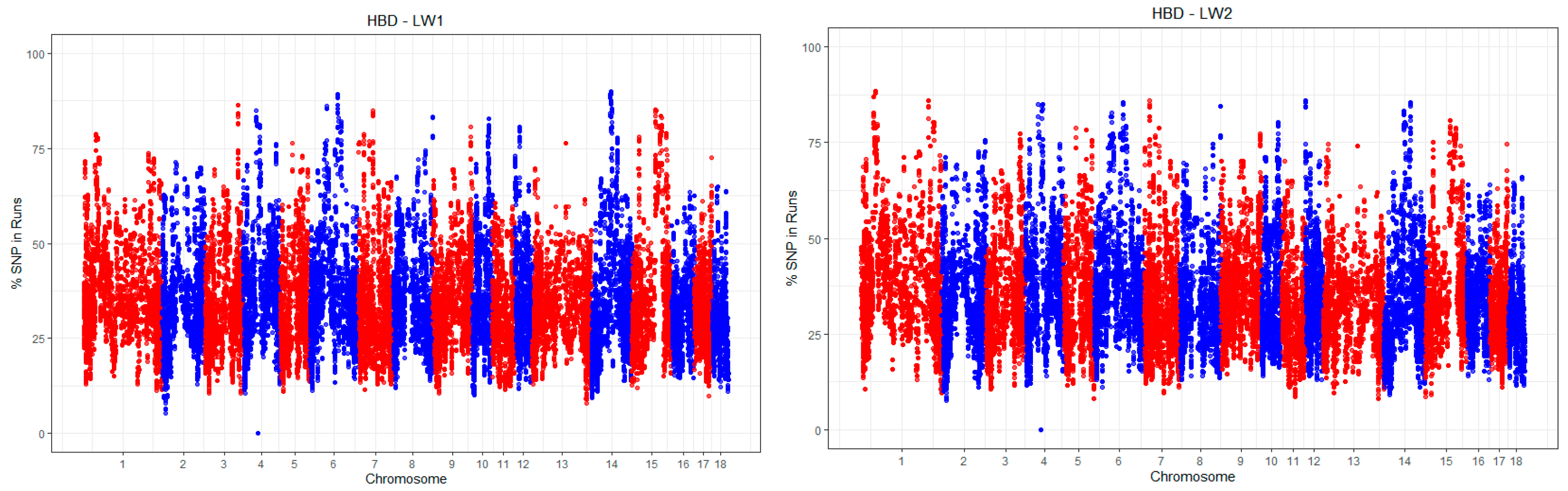
3.1. Autozygosity Analysis
3.2. Heritability of the Trait Evaluation
3.3. GWAS Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Driscoll, C.A.; Macdonald, D.W.; O’Brien, S.J. From wild animals to domestic pets, an evolutionary view of domestication. Proc. Natl. Acad. Sci. USA 2009, 106 (Suppl. S1), 9971–9978. [Google Scholar] [CrossRef]
- van Marle-Köster, E.; Visser, C. Unintended consequences of selection for increased production on the health and welfare of livestock. Arch. Anim. Breed. 2021, 64, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bakoev, S.; Kolosov, A.; Bakoev, F.; Kostyunina, O.; Bakoev, N.; Romanets, T.; Koshkina, O.; Getmantseva, L. Analysis of Homozygous-by-Descent (HBD) Segments for Purebred and Crossbred Pigs in Russia. Life 2021, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Bakoev, S.; Getmantseva, L.; Kolosova, M.; Bakoev, F.; Kolosov, A.; Romanets, E.; Shevtsova, V.; Romanets, T.; Kolosov, Y.; Usatov, A. Identifying Significant SNPs of the Total Number of Piglets Born and Their Relationship with Leg Bumps in Pigs. Biology 2024, 13, 1034. [Google Scholar] [CrossRef] [PubMed]
- Getmantseva, L.; Kolosova, M.; Bakoev, F.; Zimina, A.; Bakoev, S. Genomic regions and candi-date genes linked to capped hock in pig. Life 2021, 11, 491. [Google Scholar] [CrossRef]
- Getmantseva, L.; Kolosova, M.; Fede, K.; Korobeinikova, A.; Kolosov, A.; Romanets, E.; Bakoev, F.; Romanets, T.; Yudin, V.; Keskinov, A.; et al. Finding Predictors of Leg Defects in Pigs Using CNV-GWAS. Genes 2023, 14, 2054. [Google Scholar] [CrossRef]
- Raszek, M.M.; Guan, L.L.; Plastow, G.S. Use of genomic tools to improve cattle health in the context of infectious diseases. Front. Genet. 2016, 7, 30. [Google Scholar] [CrossRef]
- Seabury, C.M.; Oldeschulte, D.L.; Saatchi, M.; Beever, J.E.; Decker, J.E.; Halley, Y.A.; Bhattarai, E.K.; Molaei, M.; Freetly, H.C.; Hansen, S.L.; et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genom. 2017, 18, 386. [Google Scholar] [CrossRef]
- Desire, S.; Johnsson, M.; Ros-Freixedes, R.; Chen, C.Y.; Holl, J.W.; Herring, W.O.; Gorjanc, G.; Mellanby, R.J.; Hickey, J.M.; Jungnickel, M.K. A genome-wide association study for loin depth and muscle pH in pigs from intensely selected purebred lines. Genet. Sel. Evol. GSE 2023, 55, 42. [Google Scholar] [CrossRef]
- Druet, T.; Gautier, M.A. A model based approach to characterize individual inbreeding at both global and local genomic scales. Mol. Ecol. 2017, 26, 5820–5841. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220. [Google Scholar] [CrossRef] [PubMed]
- Gorssen, W.; Meyermans, R.; Buys, N.; Janssens, S. SNP genotypes reveal breed substructure, selection signatures and highly inbred regions in Piétrain pigs. Anim. Genet. 2020, 51, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, X.; Liu, J.; Zhang, W.; Zhou, M.; Wang, J.; Liu, L.; Su, S.; Zhao, F.; Chen, H.; et al. Genome-wide detection of genetic structure and runs of homozygosity analysis in Anhui indigenous and Western commercial pig breeds using PorcineSNP80k data. BMC Genom. 2022, 23, 373. [Google Scholar] [CrossRef]
- Bakoev, S.; Getmantseva, L.; Kostyunina, O.; Bakoev, N.; Prytkov, Y.; Usatov, A.; Tatarinova, T.V. Genome-wide analysis of genetic diversity and artificial selection in Large White pigs in Russia. PeerJ 2021, 9, e11595. [Google Scholar] [CrossRef]
- Kolosova, M.A.; Kolosov, A.Y.; Chernyshkov, A.S. Evaluation of the relationship between the condition of the limbs and the productive qualities of pigs. Agrar. Bull. Ural. 2024, 24, 1476–1491. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Bertrand, A.R.; Kadri, N.K.; Flori, L.; Gautier, M.; Druet, T. RZooRoH: An R package to characterize individual genomic autozygosity and identify homozygous-by-descent segments. Br. Ecol. Soc. 2019, 10, 860–866. [Google Scholar] [CrossRef]
- Schiavo, G.; Bovo, S.; Ribani, A.; Moscatelli, G.; Bonacini, M.; Prandi, M.; Mancin, E.; Mantovani, R.; Dall’Olio, S.; Fontanesi, L. Comparative analysis of inbreeding parameters and runs of homozygosity islands in 2 Italian autochthonous cattle breeds mainly raised in the Parmigiano-Reggiano cheese production region. J. Dairy Sci. 2022, 105, 2408–2425. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Yang, J.; Zaitlen, N.A.; Goddard, M.E.; Visscher, P.M.; Price, A.L. Mixed model association methods: Advantages and pitfalls. Nat. Genet. 2014, 46, 100–106. [Google Scholar] [CrossRef]
- Hinkle, R.T.; Donnelly, E.; Cody, D.B.; Sheldon, R.J.; Isfort, R.J. Activation of the vasoactive intestinal peptide 2 receptor modulates normal and atrophying skeletal muscle mass and force. J. Appl. Physiol. 2005, 98, 655–662. [Google Scholar] [CrossRef]
- Wei, Y.; Mojsov, S. Tissue specific expression of different human receptor types for pituitary adenylate cyclase activating polypeptide and vasoactive intestinal polypeptide: Implications for their role in human physiology. J. Neuroendocrinol. 1996, 8, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Asnicar, M.A.; Koster, A.; Heiman, M.L.; Tinsley, F.; Smith, D.P.; Galbreath, E.; Fox, N.; Ma, Y.L.; Blum, W.F.; Hsiung, H.M. Vasoactive intestinal polypeptide/pituitary adenylate cyclase-activating peptide receptor 2 deficiency in mice results in growth retardation and increased basal metabolic rate. Endocrinology 2002, 143, 3994–4006. [Google Scholar] [CrossRef]
- Braux, J.; Jourdain, M.L.; Guillaume, C.; Untereiner, V.; Piot, O.; Baehr, A.; Klymiuk, N.; Winter, N.; Berri, M.; Buzoni-Gatel, D.; et al. CFTR-deficient pigs display alterations of bone microarchitecture and composition at birth. J. Cyst. Fibros. 2020, 19, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Veltman, M.; De Sanctis, J.B.; Stolarczyk, M.; Klymiuk, N.; Bähr, A.; Brouwer, R.W.; Oole, E.; Shah, J.; Ozdian, T.; Liao, J.; et al. CFTR Correctors and Antioxidants Partially Normalize Lipid Imbalance but not Abnormal Basal Inflammatory Cytokine Profile in CF Bronchial Epithelial Cells. Front. Physiol. 2021, 12, 619442. [Google Scholar] [CrossRef]
- Uc, A.; Strandvik, B.; Yao, J.; Liu, X.; Yi, Y.; Sun, X.; Welti, R.; Engelhardt, J.F.; Norris, A.W. The fatty acid imbalance of cystic fibrosis exists at birth independent of feeding in pig and ferret models. Clin. Sci. 2022, 136, 1773–1791. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Guo, R.; Liu, T.; Shasaltaneh, M.D.; Wang, X.; Imani, S.; Wen, Q. Targeting Adenylate Cyclase Family: New Concept of Targeted Cancer Therapy. Front. Oncol. 2022, 12, 829212. [Google Scholar] [CrossRef]
- Cochet-Bissuel, M.; Lory, P.; Monteil, A. The sodium leak channel, NALCN, in health and disease. Front. Cell. Neurosci. 2014, 8, 132. [Google Scholar] [CrossRef]
- Tang, J.; Chen, X.; Cai, B.; Chen, G. A logical relationship for schizophrenia, bipolar, and major depressive disorder. Part 4: Evidence from chromosome 4 high-density association screen. J. Comp. Neurol. 2019, 527, 392–405. [Google Scholar] [CrossRef]
- Poirier, J.G.; Brennan, P.; McKay, J.D.; Spitz, M.R.; Bickeböller, H.; Risch, A.; Liu, G.; Le, M.L.; Tworoger, S.; McLaughlin, J.; et al. Informed genome-wide association analysis with family history as a secondary phenotype identifies novel loci of lung cancer. Genet. Epidemiol. 2015, 39, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.K.; Lea, R.A.; Holland, S.; Nguyen, Q.; Raghubar, A.M.; Sutherland, H.G.; Benton, M.C.; Haupt, L.M.; Blackburn, N.B.; Curran, J.E.; et al. Multi-phenotype genome-wide association studies of the Norfolk Island isolate implicate pleiotropic loci involved in chronic kidney disease. Sci. Rep. 2021, 11, 19425. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Feng, C.; Ma, L.; Song, C.; Wang, Y.; Da, Y.; Li, H.; Chen, K.; Ye, S.; Ge, C. Genome-wide association study of body weight in chicken F2 resource population. PLoS ONE 2011, 6, e21872. [Google Scholar] [CrossRef]
- Cha, J.; Choo, H.; Srikanth, K.; Lee, S.H.; Son, J.W.; Park, M.R.; Kim, N.; Jang, G.W.; Park, J.E. Genome-Wide Association Study Identifies 12 Loci Associated with Body Weight at Age 8 Weeks in Korean Native Chickens. Genes 2021, 12, 1170. [Google Scholar] [CrossRef] [PubMed]
- Buzanskas, M.E.; Grossi, D.A.; Ventura, R.V.; Schenkel, F.S.; Sargolzaei, M.; Meirelles, S.L.; Mokry, F.B.; Higa, R.H.; Mudadu, M.A.; da Silva, M.V.; et al. Genome-wide association for growth traits in Canchim beef cattle. PLoS ONE 2014, 9, e94802. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, Z.; Glória, L.S.; Zhang, K.; Zhang, C.; Yang, R.; Luo, X.; Jia, X.; Lai, S.J.; Chen, S.Y. Genome-Wide Association Studies for Growth Curves in Meat Rabbits Through the Single-Step Nonlinear Mixed Model. Front. Genet. 2021, 12, 750939. [Google Scholar] [CrossRef]
- Li, T.; Xing, F.; Zhang, N.; Chen, J.; Zhang, Y.; Yang, H.; Peng, S.; Ma, R.; Liu, Q.; Gan, S.; et al. Genome-Wide Association Analysis of Growth Traits in Hu Sheep. Genes 2024, 15, 1637. [Google Scholar] [CrossRef]
- Gonzaga-Jauregui, C.; Gamble, C.N.; Yuan, B.; Penney, S.; Jhangiani, S.; Muzny, D.M.; Gibbs, R.A.; Lupski, J.R.; Hecht, J.T. Mutations in COL27A1 cause Steel syndrome and suggest a founder mutation effect in the Puerto Rican population. Eur. J. Hum. Genet. EJHG 2015, 23, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.H.; Cai, J.H.; Chen, D.L.; Liao, S.H.; Lin, Y.Z.; Chung, Y.T.; Tsai, J.J.P.; Wang, C.C.N. LASSO and Bioinformatics Analysis in the Identification of Key Genes for Prognostic Genes of Gynecologic Cancer. J. Pers. Med. 2021, 11, 1177. [Google Scholar] [CrossRef]
- Qi, H.; Deng, Z.; Ye, F.; Gou, J.; Huang, M.; Xiang, H.; Li, H. Analysis of the differentially expressed genes in the combs and testes of Qingyuan partridge roosters at different developmental stages. BMC Genom. 2024, 25, 33. [Google Scholar] [CrossRef]
- Shaheen, R.; Schmidts, M.; Faqeih, E.; Hashem, A.; Lausch, E.; Holder, I.; Superti-Furga, A.; UK10K Consortium; Mitchison, H.M.; Almoisheer, A.; et al. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum. Mol. Genet. 2015, 24, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Niceta, M.; Muto, V.; Vona, B.; Pagnamenta, A.T.; Maroofian, R.; Beetz, C.; van Duyvenvoorde, H.; Dentici, M.L.; Lauffer, P.; et al. SCUBE3 loss-of-function causes a recognizable recessive developmental disorder due to defective bone morphogenetic protein signaling. Am. J. Hum. Genet. 2021, 108, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, L.G.; Zhang, L.C.; Yan, H.; Zhao, K.B.; Liang, J.; Li, N.; Pu, L.; Zhang, T.; Wang, L.X. Molecular cloning, tissue expression pattern, and copy number variation of porcine SCUBE3. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yuan, W.; Duan, S.; Jiao, K.; Zhang, Q.; Lim, E.G.; Chen, M.; Zhao, C.; Pan, P.; Liu, X.; et al. Microfluidic-Assisted Caenorhabditis elegans Sorting: Current Status and Future Prospects. Cyborg Bionic Syst. 2023, 4, 0011. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Variance | SE |
|---|---|---|
| V(G) | 0.08 | 0.02 |
| V(e) | 0.15 | 0.02 |
| Vp | 0.22 | 0.01 |
| V(G)/Vp | 0.35 | 0.08 |
| Chr | SNP | Variant | Gene |
|---|---|---|---|
| 1 | rs81351938 | intron variant | COL27A1 |
| 2 | rs81296219 | 3 prime UTR variant | CEP120 |
| 2 | rs81327279 | intron variant | PAMR1 |
| 7 | rs80937427 | intron variant | SCUBE3 |
| 8 | rs81403653 | intron variant | KCNIP4 |
| 11 | rs80793012 | intron variant | NALCN |
| 16 | rs339814189 | intron variant | ADCY2 |
| 18 | rs81297167 | intron variant | CFTR |
| 18 | rs337584442 | intron variant | MTURN |
| 18 | rs325478346 | intron variant | VIPR2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Getmantseva, L.; Kolosova, M.; Shevtsova, V.; Kolosov, A.; Bakoev, F.; Romanets, E.; Romanets, T.; Bakoev, S. Genetic Architecture of Hock Joint Bumps in Pigs: Insights from ROH and GWAS Analyses. Animals 2025, 15, 1178. https://doi.org/10.3390/ani15081178
Getmantseva L, Kolosova M, Shevtsova V, Kolosov A, Bakoev F, Romanets E, Romanets T, Bakoev S. Genetic Architecture of Hock Joint Bumps in Pigs: Insights from ROH and GWAS Analyses. Animals. 2025; 15(8):1178. https://doi.org/10.3390/ani15081178
Chicago/Turabian StyleGetmantseva, Lyubov, Maria Kolosova, Varvara Shevtsova, Anatoly Kolosov, Faridun Bakoev, Elena Romanets, Timofey Romanets, and Siroj Bakoev. 2025. "Genetic Architecture of Hock Joint Bumps in Pigs: Insights from ROH and GWAS Analyses" Animals 15, no. 8: 1178. https://doi.org/10.3390/ani15081178
APA StyleGetmantseva, L., Kolosova, M., Shevtsova, V., Kolosov, A., Bakoev, F., Romanets, E., Romanets, T., & Bakoev, S. (2025). Genetic Architecture of Hock Joint Bumps in Pigs: Insights from ROH and GWAS Analyses. Animals, 15(8), 1178. https://doi.org/10.3390/ani15081178