
Multi-Omics Data Analysis Identifies Prognostic Biomarkers across Cancers


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Analysis
2.1.1. Identification of Differentially Expressed Genes
2.1.2. Identification of Differentially Methylated Probes
2.2. Construction Gene Co-Expression & Co-Methylation Networks
| Algorithm 1: Procedure for determining pairwise gene correlations. |
| Input: expression and methylation profiles of n genes. |
| Output: pairwise gene correlations r′ij for any pair of genes i and j. |
| Compute correlation rij of each pair of genes i and j, using Pearson correlation. |
| Normalize rij for any 1 ≤ i, j ≤ n with the following steps: |
| 1. Apply Fisher’s z transformation to rij, i.e., zij |
| 2. Standardize zij, i.e., z′ij= , where and are the mean and standard deviation of zij for all 1 ≤ i, j ≤ n. |
| 3. Apply Fisher’s inverse transformation to z′ij, i.e., r′ij= |
| Return r′ij for any i, j. |
2.3. Network-Based Data Integration
2.4. Network-Based Clustering
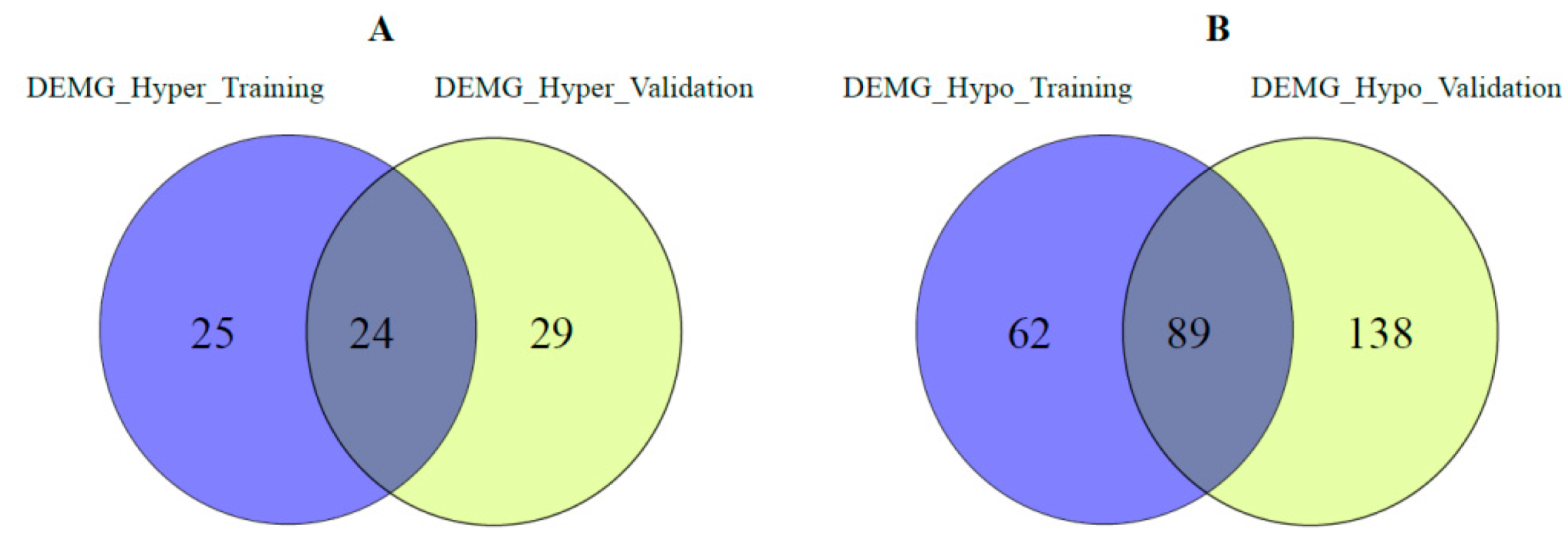
2.5. Validation Analysis
2.6. Somatic Mutation Status of Biomarkers
2.7. Survival Analysis
2.8. MOFA Analysis
3. Results
3.1. Identification of Differentially Expressed Genes/Differentially Methylated Probes
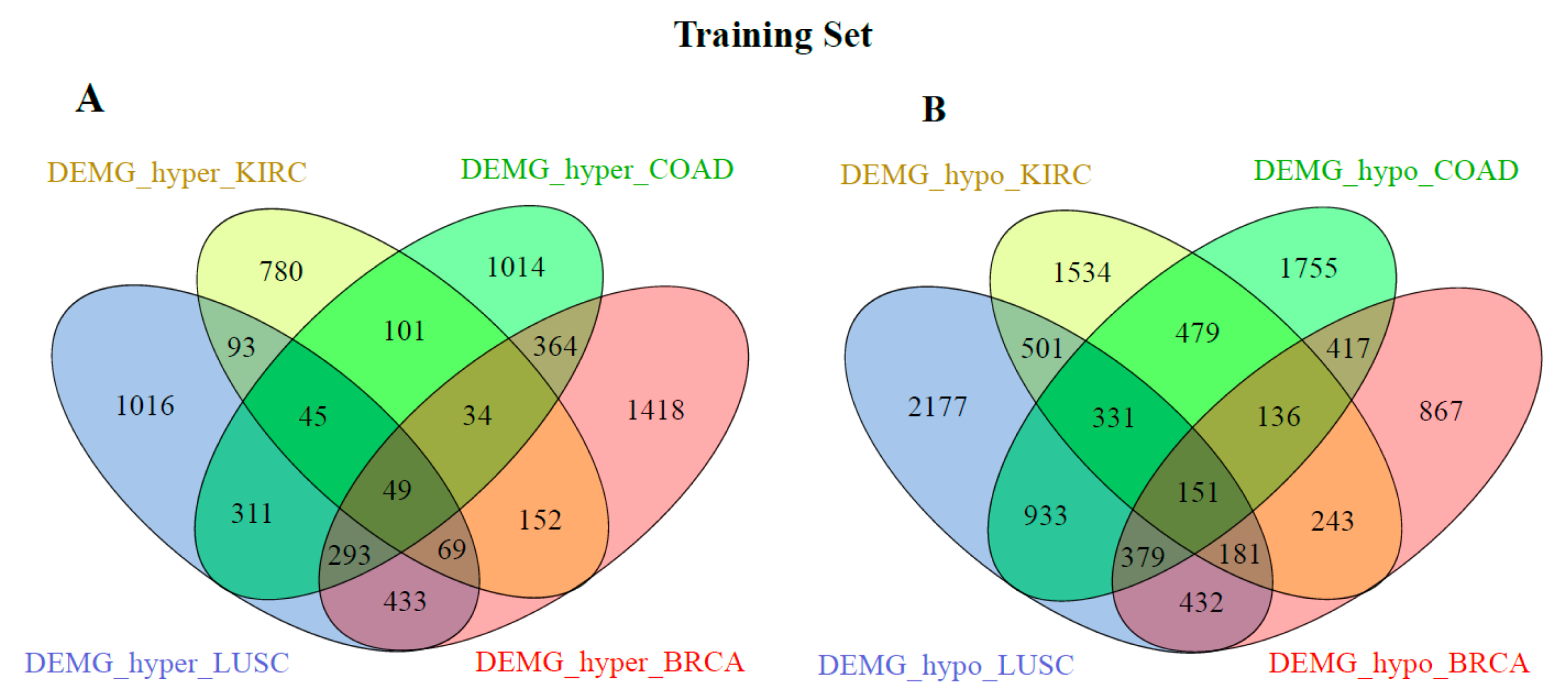
3.2. Identification of Common Genes in Different Cancer Types
3.3. Network Clustering
3.4. Somatic Mutation Status of Biomarkers
3.5. Survival Analysis
3.6. Usage of Individual Data Types for Survival Analysis
3.7. MOFA Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.; Collisson, E.; Mills, G.; Shaw, K.; Ozenberger, B.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Haas, R. Designing and Interpreting ‘Multi-Omic’Experiments That May Change Our Understanding of Biology. Curr. Opin. Syst. Biol. 2017, 6, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chaudhary, K.; Garmire, L. More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front. Genet. 2017, 8, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhang, S. Tumor Characterization and Stratification By Integrated Molecular Profiles Reveals Essential Pan-Cancer Features. BMC Genom. 2015, 16, 503. [Google Scholar] [CrossRef] [Green Version]
- Cantini, L.; Medico, E.; Fortunato, S.; Caselle, M. Detection of Gene Communities in Multi-Networks Reveals Cancer Drivers. Sci. Rep. 2015, 5, 17386. [Google Scholar] [CrossRef] [Green Version]
- Nicora, G.; Vitali, F.; Dagliati, A.; Geifman, N.; Bellazzi, R. Integrated Multi-Omics Analyses in Oncology: A Review of Machine Learning Methods and Tools. Front. Oncol. 2020, 10, 1030. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-Omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 117793221989905. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, T.; Jeong, H.; Sohn, K. Integrative Pathway-Based Survival Prediction Utilizing the Interaction between Gene Expression and DNA Methylation in Breast Cancer. BMC Med. Genom. 2018, 11, 33–43. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, B.; Lin, T.; Zhang, Y.; Zhang, L.; Wang, M. Multiomics Analysis on DNA Methylation and The Expression of Both Messenger RNA and Microrna in Lung Adenocarcinoma. J. Cell. Physiol. 2018, 234, 7579–7586. [Google Scholar] [CrossRef]
- Huo, X.; Sun, H.; Cao, D.; Yang, J.; Peng, P.; Yu, M.; Shen, K. Identification of Prognosis Markers for Endometrial Cancer By Integrated Analysis of DNA Methylation and RNA-Seq Data. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, N.; Wu, Y.; Ke, Z.; Liang, Y.; Cai, H.; Su, W.; Tao, X.; Chen, S.; Zheng, Q.; Wei, Y.; et al. Identification of Key DNA Methylation-Driven Genes in Prostate Adenocarcinoma: An Integrative Analysis of TCGA Methylation Data. J. Transl. Med. 2019, 17, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Wang, F.; Meng, Z.; Wang, N.; Zhou, C.; Zhang, J.; Zhao, L.; Wang, G.; Shan, B. Uncovering Potential Genes in Colorectal Cancer Based on Integrated and DNA Methylation Analysis in The Gene Expression Omnibus Database. BMC Cancer 2022, 22, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, M.; Zhang, F.; Kong, X. An Integrated Analysis of Genome-Wide DNA Methylation and Gene Expression Data in Hepatocellular Carcinoma. FEBS Open Bio 2018, 8, 1093–1103. [Google Scholar] [CrossRef]
- Champion, M.; Brennan, K.; Croonenborghs, T.; Gentles, A.; Pochet, N.; Gevaert, O. Module Analysis Captures Pancancer Genetically and Epigenetically Deregulated Cancer Driver Genes for Smoking and Antiviral Response. EBioMedicine 2018, 27, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Dimitrakopoulos, C.; Hindupur, S.; Häfliger, L.; Behr, J.; Montazeri, H.; Hall, M.; Beerenwinkel, N. Network-Based Integration of Multi-Omics Data for Prioritizing Cancer Genes. Bioinformatics 2018, 34, 2441–2448. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Mezlini, A.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity Network Fusion for Aggregating Data Types on A Genomic Scale. Nat. Methods 2014, 11, 333–337. [Google Scholar] [CrossRef]
- Tian, Z.; Guo, M.; Wang, C.; Xing, L.; Wang, L.; Zhang, Y. Constructing an Integrated Gene Similarity Network for The Identification of Disease Genes. J. Biomed. Semant. 2017, 8, 32. [Google Scholar] [CrossRef]
- Pidò, S.; Ceddia, G.; Masseroli, M. Computational Analysis of Fused Co-Expression Networks for The Identification of Candidate Cancer Gene Biomarkers. npj Syst. Biol. Appl. 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Tanvir, R.B.; Maharjan, M.; Mondal, A.M. Community Based Cancer Biomarker Identification from Gene Co-Expression Network. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; p. 545. [Google Scholar]
- Wang, Y.; Liu, Z. Identifying Biomarkers for Breast Cancer by Gene Regulatory Network Rewiring. BMC Bioinform. 2021, 22, 1–14. [Google Scholar] [CrossRef]
- Yu, L.; Huang, Q.; Zhou, X. Identification of Cancer Hallmarks Based on The Gene Co-Expression Networks of Seven Cancers. Front. Genet. 2019, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Marcucci, G.; Yan, P.; Maharry, K.; Frankhouser, D.; Nicolet, D.; Metzeler, K.; Kohlschmidt, J.; Mrózek, K.; Wu, Y.; Bucci, D.; et al. Epigenetics Meets Genetics in Acute Myeloid Leukemia: Clinical Impact of a Novel Seven-Gene Score. J. Clin. Oncol. 2014, 32, 548–556. [Google Scholar] [CrossRef]
- Hu, C.; Zhou, Y.; Liu, C.; Kang, Y. A Novel Scoring System for Gastric Cancer Risk Assessment Based on The Expression of Three CLIP4 DNA Methylation-Associated Genes. Int. J. Oncol. 2018, 53, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Kaur, H.; Dhall, A.; Kumar, R.; Raghava, G. Identification of Platform-Independent Diagnostic Biomarker Panel for Hepatocellular Carcinoma Using Large-Scale Transcriptomics Data. Front. Genet. 2020, 10, 1306. [Google Scholar] [CrossRef]
- The International Cancer Genome Consortium. International Network of Cancer Genome Projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [Green Version]
- GDC. Available online: https://portal.gdc.cancer.gov/ (accessed on 20 February 2022).
- Benjamini, Y.; Hochberg, Y. Controlling The False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Silva, T.; Coetzee, S.; Gull, N.; Yao, L.; Hazelett, D.; Noushmehr, H.; Lin, D.; Berman, B. ELMER V.2: An R/Bioconductor Package to Reconstruct Gene Regulatory Networks from DNA Methylation and Transcriptome Profiles. Bioinformatics 2018, 35, 1974–1977. [Google Scholar] [CrossRef] [Green Version]
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human. 2019. Available online: http://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html (accessed on 1 May 2023).
- Li, W.; Liu, C.; Zhang, T.; Li, H.; Waterman, M.; Zhou, X. Integrative Analysis of Many Weighted Co-Expression Networks Using Tensor Computation. PLoS Comput. Biol. 2011, 7, 1001106. [Google Scholar] [CrossRef] [Green Version]
- Csárdi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. Inter J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Clauset, A.; Newman, M.; Moore, C. Finding Community Structure in Very Large Networks. Phys. Rev. E 2004, 70, 066111. [Google Scholar] [CrossRef] [Green Version]
- Rosvall, M.; Bergstrom, C. Maps of Random Walks on Complex Networks Reveal Community Structure. Proc. Natl. Acad. Sci. USA 2008, 105, 1118–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blondel, V.; Guillaume, J.; Lambiotte, R.; Lefebvre, E. Fast Unfolding of Communities in Large Networks. J. Stat. Mech. Theory Exp. 2008, 10, 10008. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Datta, S. Methods for Evaluating Clustering Algorithms for Gene Expression Data Using a Reference Set of Functional Classes. BMC Bioinform. 2006, 7, 397. [Google Scholar] [CrossRef] [PubMed]
- Bruno, G.; Fiori, A.M. Microarray Clustering Analysis. J. Parallel Distrib. Comput. 2013, 73, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Sheng, Z.; Ma, C.; Tang, K.; Zhu, R.; Wu, Z.; Shen, R.; Feng, J.; Wu, D.; Huang, D.; et al. Combining Genomic and Network Characteristics for Extended Capability in Predicting Synergistic Drugs for Cancer. Nat. Commun. 2015, 6, 8481. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.A. On the Interpretation of Χ2 from Contingency Tables, and the Calculation of P. J. R Stat. Soc. 1922, 85, 87–94. [Google Scholar] [CrossRef]
- Cox, D.R.; Oakes, D. Analysis of Survival Data, 1st ed.; Chapman and Hall/CR: Boca Raton, FL, USA, 1984. [Google Scholar]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Argelaguet, R.; Velten, B.; Arnol, D.; Dietrich, S.; Zenz, T.; Marioni, J.C.; Buettner, F.; Huber, W.; Stegle, O. Multi-Omics Factor Analysis-a Framework for Unsupervised Integration of Multi-Omics Data Sets. Mol. Syst. Biol. 2018, 14, e8124. [Google Scholar] [CrossRef]
- Fan, S.; Tang, J.; Li, N.; Zhao, Y.; Ai, R.; Zhang, K.; Wang, M.; Du, W.; Wang, W. Integrative Analysis with Expanded DNA Methylation Data Reveals Common Key Regulators and Pathways in Cancers. Npj Genom. Med. 2019, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Mo, Q.; Wang, S.; Seshan, V.; Olshen, A.; Schultz, N.; Sander, C.; Powers, R.; Ladanyi, M.; Shen, R. Pattern Discovery and Cancer Gene Identification in Integrated Cancer Genomic Data. Proc. Natl. Acad. Sci. USA 2013, 110, 4245–4250. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Zhou, B.; Chen, J.; Hu, W.; Bai, R.; Ye, C.; Weng, X.; Zheng, S. Significant Prognostic Values of Differentially Expressed-Aberrantly Methylated Hub Genes in Breast Cancer. J. Cancer 2019, 10, 6618–6634. [Google Scholar] [CrossRef] [Green Version]
- Le, H.; Nguyen, V.M.; Nguyen, Q.H.; Le, D.H. A Biphasic Deep Semi-Supervised Framework for Subtype Classification and Biomarker Discovery. bioRxiv 2022. [Google Scholar]
- Fiorentino, G.; Visintainer, R.; Domenici, E.; Lauria, M.; Marchetti, L.M.O.U.S.S.E. Multi-Omics Using Subject-Specific Signatures. Cancers 2021, 13, 3423. [Google Scholar] [CrossRef]
- Sheng, H.; Pan, H.; Yao, M.; Xu, L.; Lu, J.; Liu, B.; Shen, J.; Shen, H. Integrated Analysis of Circular RNA-Associated Cerna Network Reveals Potential Circrna Biomarkers in Human Breast Cancer. Comput. Math. Methods Med. 2021, 2021, 1732176. [Google Scholar] [CrossRef]
- Shi, K.; Gao, L.; Wang, B. Discovering Potential Cancer Driver Genes By an Integrated Network-Based Approach. Mol. BioSystems 2016, 12, 2921–2931. [Google Scholar] [CrossRef]
- Hua, P.; Zhang, Y.; Jin, C.; Zhang, G.; Wang, B. Integration of gene profile to explore the hub genes of lung adenocarcinoma: A quasi-experimental study. Medicine 2020, 99, 22727. [Google Scholar] [CrossRef]
- Shi, K.; Li, N.; Yang, M.; Li, W. Identification of Key Genes and Pathways in Female Lung Cancer Patients Who Never Smoked by a Bioinformatics Analysis. J. Cancer 2019, 10, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Buttarelli, M.; Ciucci, A.; Palluzzi, F.; Raspaglio, G.; Marchetti, C.; Perrone, E.; Minucci, A.; Giacò, L.; Fagotti, A.; Scambia, G.; et al. Identification of a Novel Gene Signature Predicting Response to First-Line Chemotherapy in BRCA Wild-Type High-Grade Serous Ovarian Cancer Patients. J. Exp. Clin. Cancer Res. CR 2022, 41, 50. [Google Scholar] [CrossRef]
- Jiang, M.M.; Zhao, F.; Lou, T.T. Assessment of Significant Pathway Signaling and Prognostic Value of GNG11 in Ovarian Serous Cystadenocarcinoma. Int. J. Gen. Med. 2021, 14, 2329–2341. [Google Scholar] [CrossRef]
- Zhang, X.; Kang, X.; Jin, L. ABCC9, NKAPL, and TMEM132C Are Potential Diagnostic and Prognostic Markers in Triple-Negative Breast Cancer. Cell Biol. Int. 2020, 44, 2002–2010. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Wang, Y.; Hu, K.; Wang, F.; Sun, T.; Li, Q. WGCNA Reveals Key Gene Modules Regulated by the Combined Treatment of Colon Cancer with PHY906 and CPT11. Biosci. Rep. 2020, 40, 20200935. [Google Scholar] [CrossRef] [PubMed]
- Clermont, P.; Crea, F.; Chiang, Y.; Lin, D.; Zhang, A.; Wang, J.; Parolia, A.; Wu, R.; Xue, H.; Wang, Y.; et al. Identification of The Epigenetic Reader CBX2 As A Potential Drug Target in Advanced Prostate Cancer. Clin. Epigenetics 2016, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, J.; Tian, Y.; Wang, C.; Jiang, K.; Li, R.; Yao, Y.; Zhang, R.; Sun, D.; Liang, R.; Gao, Z.; et al. CBX2 Regulates Proliferation and Apoptosis Via The Phosphorylation of YAP in Hepatocellular Carcinoma. J. Cancer 2019, 10, 2706–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, L.; Watson, Z.; Qamar, L.; Yamamoto, T.; Post, M.; Berning, A.; Spillman, M.; Behbakht, K.; Bitler, B. CBX2 Identified as Driver of Anoikis Escape and Dissemination in High Grade Serous Ovarian Cancer. Oncogenesis 2018, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Chen, H.; Duan, Y.; Lan, B.; Liu, C.; Hu, H.; Dong, X.; Zhang, Q.; Cheng, Y.; Liu, M.; et al. CBX2 and EZH2 Cooperatively Promote The Growth and Metastasis of Lung Adenocarcinoma. Mol. Ther. Nucleic Acids 2022, 27, 670–684. [Google Scholar] [CrossRef]
- Ma, T.; Ma, N.; Chen, J.; Tang, F.; Zong, Z.; Yu, Z.; Chen, S.; Zhou, T. Expression and Prognostic Value of Chromobox Family Members in Gastric Cancer. J. Gastrointest. Oncol. 2020, 11, 983–998. [Google Scholar] [CrossRef]
- Li, Q.; Pan, Y.; Cao, Z.; Zhao, S. Comprehensive Analysis of Prognostic Value and Immune Infiltration of Chromobox Family Members in Colorectal Cancer. Front. Oncol. 2020, 10, 582667. [Google Scholar] [CrossRef]
- Zhou, H.; Xiong, Y.; Liu, Z.; Hou, S.; Zhou, T. Expression and Prognostic Significance of CBX2 in Colorectal Cancer: Database Mining for CBX Family Members in Malignancies and Vitro Analyses. Cancer Cell Int. 2021, 21, 1–6. [Google Scholar]
- Bilton, L.; Warren, C.; Humphries, R.; Kalsi, S.; Waters, E.; Francis, T.; Dobrowinski, W.; Beltran-Alvarez, P.; Wade, M. The Epigenetic Regulatory Protein CBX2 Promotes Mtorc1 Signalling and Inhibits DREAM Complex Activity to Drive Breast Cancer Cell Growth. Cancers 2022, 14, 3491. [Google Scholar] [CrossRef]
- Zheng, S.; Lv, P.; Su, J.; Miao, K.; Xu, H.; Li, M. Overexpression of CBX2 in Breast Cancer Promotes Tumor Progression through the PI3K/AKT Signaling Pathway. Am. J. Transl. Res. 2019, 11, 1668–1682. [Google Scholar]
- Li, X.; Gou, J.; Li, H.; Yang, X. Bioinformatic Analysis of The Expression and Prognostic Value of Chromobox Family Proteins in Human Breast Cancer. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Piqué, D.; Montagna, C.; Greally, J.; Mar, J. A Novel Approach To Modelling Transcriptional Heterogeneity Identifies The Oncogene Candidate CBX2 in Invasive Breast Carcinoma. Br. J. Cancer 2019, 120, 746–753. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. Cell Cycle, Cdks and Cancer: A Changing Paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Hannon, G.; Casso, D.; Beach, D. KAP: A Dual Specificity Phosphatase That Interacts with Cyclin-Dependent Kinases. Proc. Natl. Acad. Sci. USA 1994, 91, 1731–1735. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Tawab, M.; Fouad, H.; Othman, A.; Eid, R.; Mohammed, M.; Hassan, A. Reyad Evaluation of Gene Expression of PLEKHS1, AADAC, and CDKN3 As Novel Genomic Markers in Gastric Carcinoma. PLoS ONE 2022, 17, 0265184. [Google Scholar] [CrossRef]
- Li, Y.; Ji, S.; Fu, L.; Jiang, T.; Wu, D.; Meng, F. Knockdown of Cyclin-Dependent Kinase Inhibitor 3 Inhibits Proliferation and Invasion in Human Gastric Cancer Cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2017, 25, 721–731. [Google Scholar] [CrossRef]
- Chang, S.; Chen, T.; Lee, Y.; Lee, S.; Lin, L.; He, H. CDKN3 Expression Is an Independent Prognostic Factor and Associated with Advanced Tumor Stage in Nasopharyngeal Carcinoma. Int. J. Med. Sci. 2018, 15, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Chen, L.; Huang, Q.; Shen, T.; Welsh, E.; Teer, J.; Cai, J.; Cress, W.; Wu, J. Overexpression of Major CDKN3 Transcripts Is Associated with Poor Survival in Lung Adenocarcinoma. Br. J. Cancer 2015, 113, 1735–1743. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Huang, X.; Shao, K.; Li, G.; Wang, J.; Yang, H.; Hou, Y. Integrated Bioinformatics Analysis To Identify 15 Hub Genes in Breast Cancer. Oncol. Lett. 2019, 18, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Che, N.; Jin, Y.; Li, J.; Yang, W. CDKN3 Overcomes Bladder Cancer Cisplatin Resistance Via LDHA-Dependent Glycolysis Reprogramming. OncoTargets Ther. 2022, 15, 299–311. [Google Scholar] [CrossRef]
- Barrón, E.; Roman-Bassaure, E.; Sánchez-Sandoval, A.; Espinosa, A.; Guardado-Estrada, M.; Medina, I.; Juárez, E.; Alfaro, A.; Bermúdez, M.; Zamora, R.; et al. CDKN3 Mrna As A Biomarker for Survival and Therapeutic Target in Cervical Cancer. PLoS ONE 2015, 10, 0137397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Liao, K.; Guo, H.; Zhou, S.; Yu, R.; Liu, Y.; Pan, Y.; Pu, J. Integrated Transcriptomics Explored The Cancer-Promoting Genes CDKN3 in Esophageal Squamous Cell Cancer. J. Cardiothorac. Surg. 2021, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sun, J. Mechanistic Studies of Cyclin-Dependent Kinase Inhibitor 3 (CDKN3) in Colorectal Cancer. Asian Pac. J. Cancer Prev. 2015, 16, 965–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.H.; Zhang, L.; Wu, Y.H. CDKN3 Regulates Cisplatin Resistance to Colorectal Cancer through TIPE1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3614–3623. [Google Scholar]
- Joseph, J.; Radulovich, N.; Wang, T. Rho guanine nucleotide exchange factor ARHGEF10 is a putative tumor suppressor in pancreatic ductal adenocarcinoma. Oncogene 2020, 39, 308–321. [Google Scholar] [CrossRef]
- Xue, W.; Kitzing, T.; Roessler, S.; Zuber, J.; Krasnitz, A.; Schultz, N.; Revill, K.; Weissmueller, S.; Rappaport, A.R.; Simon, J.; et al. A Cluster of Cooperating Tumor-Suppressor Gene Candidates in Chromosomal Deletions. Proc. Natl. Acad. Sci. USA 2012, 109, 8212–8217. [Google Scholar] [CrossRef] [Green Version]
- Cooke, S.L.; Pole, J.C.; Chin, S.F.; Ellis, I.O.; Caldas, C.; Edwards, P.A. High-Resolution Array CGH Clarifies Events Occurring on 8p in Carcinogenesis. BMC Cancer 2008, 8, 288. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.V.; Taylor, C.; Platt, F.; Hurst, C.D.; Aveyard, J.; Knowles, M.A. Mutation and Homozygous Deletion of ARHGEF10 in Bladder Cancer; a Candidate Tumour Suppressor Gene at 8p23. 3. Cancer Genet. Cytogenet. 2010, 203, 68. [Google Scholar] [CrossRef]
- Ranta, S.; Zhang, Y.; Ross, B. The Neuronal Ceroid Lipofuscinoses in Human EPMR and Mnd Mutant Mice Are Associated with Mutations in CLN8. Nat. Genet. 1999, 23, 233–236. [Google Scholar] [CrossRef]
- Yap, S.Q.; Mathavarajah, S.; Huber, R.J. The Converging Roles of Batten Disease Proteins in Neurodegeneration and Cancer. iScience 2021, 24, 102337. [Google Scholar] [CrossRef]
- Zimmermann, R.; Müller, L.; Wullich, B. Protein Transport Into The Endoplasmic Reticulum: Mechanisms and Pathologies. Trends Mol. Med. 2006, 12, 567–573. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, F.; Peng, X.; Li, X.; Luo, N.; Zhu, W.; Fu, M.; Li, Q.; Hu, G. Hypoxia Constructing The Prognostic Model of Colorectal Adenocarcinoma and Related To The Immune Microenvironment. Front. Cell Dev. Biol. 2021, 9, 665364. [Google Scholar] [CrossRef]
- Ma, J.; He, Z.; Zhang, H.; Zhang, W.; Gao, S.; Ni, X. SEC61G Promotes Breast Cancer Development and Metastasis Via Modulating Glycolysis and Is Transcriptionally Regulated By E2F1. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Jin, L.; Chen, D.; Hirachan, S.; Bhandari, A.; Huang, Q. SEC61G Regulates Breast Cancer Cell Proliferation and Metastasis By Affecting The Epithelial-Mesenchymal Transition. J. Cancer 2022, 13, 831–846. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, L.; Zang, Y.; Xu, Z. Identification of Core Genes and Key Pathways Via Integrated Analysis of Gene Expression and DNA Methylation Profiles in Bladder Cancer. Med. Sci. Monit. 2018, 24, 3024–3033. [Google Scholar] [CrossRef]
- Meng, H.; Jiang, X.; Wang, J.; Sang, Z.; Guo, L.; Yin, G.; Wang, Y. SEC61G Is Upregulated and Required for Tumor Progression in Human Kidney Cancer. Mol. Med. Rep. 2021, 23, 427. [Google Scholar] [CrossRef]
- Gao, H.; Niu, W.; He, Z.; Gao, C.; Peng, C.; Niu, J. SEC61G Plays an Oncogenic Role in Hepatocellular Carcinoma Cells. Cell Cycle 2020, 19, 3348–3361. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Y.; Gong, X.; Guo, Q.; Lin, C.; Luo, Q.; Tu, Z.; Pan, J.; Li, J. SEC61G Overexpression and DNA Amplification Correlates with Prognosis and Immune Cell Infiltration in Head and Neck Squamous Cell Carcinoma. Cancer Med. 2021, 10, 7847–7862. [Google Scholar] [CrossRef]
- Liu, B.; Liu, J.; Liao, Y.; Jin, C.; Zhang, Z.; Zhao, J.; Liu, K.; Huang, H.; Cao, H.; Cheng, Q. Identification of SEC61G as a Novel Prognostic Marker for Predicting Survival and Response to Therapies in Patients with Glioblastoma. Med. Sci. Monit. 2019, 25, 3624–3635. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, Z.; Zhang, M.; Yu, Y.; Chen, R.; Lu, T.; Liu, L.; Ma, J.; Liu, T.; Zheng, H.; et al. Prognostic Value of SEC61G in Lung Adenocarcinoma: A Comprehensive Study Based on Bioinformatics and In Vitro Validation. BMC Cancer 2021, 21, 1216. [Google Scholar] [CrossRef]
- Cheng, C.; Wang, T.; Chen, P.; Wu, W.; Lai, J.; Chang, P.; Hong, Y.; Huang, C.; Wang, F. Computer-Aided Design for Identifying Anticancer Targets in Genome-Scale Metabolic Models of Colon Cancer. Biology 2021, 10, 1115. [Google Scholar] [CrossRef] [PubMed]
- Sekar, D.; Dillmann, C.; Sirait-Fischer, E.; Fink, A.; Zivkovic, A.; Baum, N.; Strack, E.; Klatt, S.; Zukunft, S.; Wallner, S.; et al. Phosphatidylserine Synthase PTDSS1 Shapes The Tumor Lipidome to Maintain Tumor-Promoting Inflammation. Cancer Res. 2022, 82, 1617–1632. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Hui, R.; Nouws, J.; Sauler, M.; Zeng, T.; Wu, Q. Untargeted Metabolomics Analysis of Esophageal Squamous Cell Cancer Progression. J. Transl. Med. 2022, 20, 127. [Google Scholar] [CrossRef]
- N’Guessan, K.; Davis, H.; Chu, Z.; Vallabhapurapu, S.; Lewis, C.; Franco, R.; Olowokure, O.; Ahmad, S.; Yeh, J.; Bogdanov, V.; et al. Enhanced Efficacy of Combination of Gemcitabine and Phosphatidylserine-Targeted Nanovesicles against Pancreatic Cancer. Mol. Ther. 2020, 28, 1876–1886. [Google Scholar] [CrossRef]
- Li, M.; Xu, D.; Lin, S.; Yang, Z.; Xu, T.; Yang, J.; Lin, Z.; Huang, Z. Transcriptional Expressions of Hsa-Mir-183 Predicted Target Genes As Independent Indicators for Prognosis in Bladder Urothelial Carcinoma. Aging 2022, 14, 3782–3800. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, M.; Chen, W.; Wu, W.; Wang, F. Optimization of A Modeling Platform To Predict Oncogenes from Genome-Scale Metabolic Networks of Non-Small-Cell Lung Cancers. FEBS Open Bio 2021, 11, 2078–2094. [Google Scholar] [CrossRef]
- Cheng, C.; Hua, J.; Tan, J.; Qian, W.; Zhang, L.; Hou, X. Identification of Differentially Expressed Genes, Associated Functional Terms Pathways, and Candidate Diagnostic Biomarkers in Inflammatory Bowel Diseases by Bioinformatics Analysis. Exp. Ther. Med. 2019, 18, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Moradi, S.; Tapak, L.; Afshar, S. Identification of Novel Noninvasive Diagnostics Biomarkers in the Parkinson’s Diseases and Improving the Disease Classification Using Support Vector Machine. BioMed Res. Int. 2022, 2022, 5009892. [Google Scholar] [CrossRef]
- Nolte, I.M.; Munoz, M.L.; Tragante, V.; Amare, A.T.; Jansen, R.; Vaez, A.; von der Heyde, B.; Avery, C.L.; Bis, J.C.; Dierckx, B.; et al. Genetic Loci Associated with Heart Rate Variability and Their Effects on Cardiac Disease Risk. Nat. Commun. 2017, 8, 15805. [Google Scholar] [CrossRef] [Green Version]
- Ou, H.; Fan, Y.; Guo, X.; Lao, Z.; Zhu, M.; Li, G.; Zhao, L. Identifying Key Genes Related to Inflammasome in Severe COVID-19 Patients Based on a Joint Model with Random Forest and Artificial Neural Network. Front. Cell. Infect. Microbiol. 2023, 13, 1139998. [Google Scholar] [CrossRef]
- Yue, Q.; Li, Z.; Zhang, Q.; Jin, Q.; Zhang, X.; Jin, G. Identification of Novel Hub Genes Associated with Psoriasis Using Integrated Bioinformatics Analysis. Int. J. Mol. Sci. 2022, 23, 15286. [Google Scholar] [CrossRef]
- Yao, S.; Deng, M.; Du, X.; Huang, R.; Chen, Q. A Novel Hypoxia Related Marker in Blood Link to Aid Diagnosis and Therapy in Osteoarthritis. Genes 2022, 13, 1501. [Google Scholar] [CrossRef]
- Verhoeven, K.; De Jonghe, P.; Van de Putte, T.; Nelis, E.; Zwijsen, A.; Verpoorten, N.; De Vriendt, E.; Jacobs, A.; Van Gerwen, V.; Francis, A.; et al. Slowed Conduction and Thin Myelination of Peripheral Nerves Associated with Mutant Rho Guanine-Nucleotide Exchange Factor 10. Am. J. Hum. Genet. 2003, 73, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Park, M.J.; Park, S.; Cheng, J.Y.; Lee, E.-S. Differential Expression of Novel Genes and Signalling Pathways of Senescent CD8+ T Cell Subsets in Behçet’s Disease. Clin. Exp. Rheumatol. 2020, 38 (Suppl. S127), 17–25. [Google Scholar]
- Zhang, C.K.; Stein, P.B.; Liu, J.; Wang, Z.; Yang, R.; Cho, J.H.; Gregersen, P.K.; Aerts, J.M.F.G.; Zhao, H.; Pastores, G.M.; et al. Genome-Wide Association Study of N370S Homozygous Gaucher Disease Reveals the Candidacy of CLN8 Gene as a Genetic Modifier Contributing to Extreme Phenotypic Variation. Am. J. Hematol. 2012, 87, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Sahin, Y.; Güngör, O.; Gormez, Z.; Demirci, H.; Ergüner, B.; Güngör, G.; Dilber, C. Exome Sequencing Identifies a Novel Homozygous CLN8 Mutation in a Turkish Family with Northern Epilepsy. Acta Neurol. Belg 2017, 117, 159–167. [Google Scholar] [CrossRef]
- Norling, A.; Hirschberg, A.L.; Iwarsson, E.; Wedell, A.; Barbaro, M. CBX2 Gene Analysis in Patients with 46,XY and 46,XX Gonadal Disorders of Sex Development. Fertil. Steril. 2013, 99, 819–826.e3. [Google Scholar] [CrossRef]
- He, H.; Yang, Y.; Wang, L.; Guo, Z.; Ye, L.; Ou-Yang, W.; Yang, M. Combined Analysis of Single-Cell and Bulk RNA Sequencing Reveals the Expression Patterns of Circadian Rhythm Disruption in the Immune Microenvironment of Alzheimer’s Disease. Front. Immunol. 2023, 14, 1182307. [Google Scholar] [CrossRef]
- Staneva, R.; Rukova, B.; Hadjidekova, S.; Nesheva, D.; Antonova, O.; Dimitrov, P.; Simeonov, V.; Stamenov, G.; Cukuranovic, R.; Cukuranovic, J.; et al. Whole Genome Methylation Array Analysis Reveals New Aspects in Balkan Endemic Nephropathy Etiology. BMC Nephrol. 2013, 14, 225. [Google Scholar] [CrossRef] [Green Version]
- Krzyzewska, I.M.; Lauffer, P.; Mul, A.N.; van der Laan, L.; Yim, A.Y.F.L.; Cobben, J.M.; Niklinski, J.; Chomczyk, M.A.; Smigiel, R.; Mannens, M.M.A.M.; et al. Expression Quantitative Trait Methylation Analysis Identifies Whole Blood Molecular Footprint in Fetal Alcohol Spectrum Disorder (FASD). Int. J. Mol. Sci. 2023, 24, 6601. [Google Scholar] [CrossRef]
- Tamhankar, P.M.; Vasudevan, L.; Bansal, V.; Menon, S.R.; Gawde, H.M.; D’Souza, A.; Babu, S.; Kondurkar, S.; Adhia, R.; Das, D.K. Lenz-Majewski Syndrome: Report of a Case with Novel Mutation in PTDSS1 Gene. Eur. J. Med. Genet. 2015, 58, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Soueid, J.; Kourtian, S.; Makhoul, N.J.; Makoukji, J.; Haddad, S.; Ghanem, S.S.; Kobeissy, F.; Boustany, R.-M. RYR2, PTDSS1 and AREG Genes Are Implicated in a Lebanese Population-Based Study of Copy Number Variation in Autism. Sci. Rep. 2016, 6, 19088. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Numberof Training Samples | Numberof Validation Samples | ||
|---|---|---|---|---|
| Tumor | Normal | Tumor | Normal | |
| COAD | 74 | 19 | 78 | 19 |
| KIRC | 90 | 24 | 91 | 24 |
| BRCA | 261 | 83 | 279 | 83 |
| LUSC | 153 | 7 | 152 | 7 |
| Cancer Type | DEMG_Common |
|---|---|
| Brca_hypo | 2428 |
| Lusc_hypo | 3235 |
| Coad_hypo | 3382 |
| Kırc_hypo | 3184 |
| Brca_hyper | 2288 |
| Lusc_hyper | 1749 |
| Coad_hyper | 1475 |
| Kırc_hyper | 1063 |
| Cancer Type | Number of Differentially Methylated Probes | Number of Nearby Genes | Number of Probe-Gene Pairs | |||
|---|---|---|---|---|---|---|
| Hypo-M | Hyper-M | Hypo-M | Hyper-M | Hypo-M | Hyper-M | |
| COAD | 3103 | 2195 | 62,039 | 43,895 | 2561 | 6117 |
| KIRC | 1277 | 691 | 25,540 | 13,820 | 2388 | 2277 |
| BRCA | 1252 | 1048 | 25,040 | 20,953 | 2490 | 4606 |
| LUSC | 3415 | 1949 | 68,300 | 38,980 | 2588 | 3451 |
| Cancer Type | Number of Differentially Methylated Probes | Number of Nearby Genes | Number of Probe-Gene Pairs | |||
|---|---|---|---|---|---|---|
| Hypo-M | Hyper-M | Hypo-M | Hyper-M | Hypo-M | Hyper-M | |
| COAD | 3084 | 1729 | 61,666 | 34,580 | 5324 | 3615 |
| KIRC | 1809 | 780 | 36,180 | 15,600 | 3440 | 2458 |
| BRCA | 1436 | 1278 | 28,720 | 18,180 | 2925 | 4225 |
| LUSC | 2121 | 1957 | 42,420 | 39,140 | 1543 | 4737 |
| Cancer Type | DEG | DMG_Hypo | DMG_Hyper | DEMG_Hypo | DEMG_Hyper |
|---|---|---|---|---|---|
| COAD | 10,916 | 10,676 | 5012 | 4581 | 2211 |
| KIRC | 12,273 | 7005 | 2524 | 3556 | 1323 |
| BRCA | 14,294 | 4971 | 4773 | 2806 | 2812 |
| LUSC | 11,585 | 10,898 | 4666 | 5085 | 2309 |
| Cancer Type | DEG | DMG_Hypo | DMG_Hyper | DEMG_Hypo | DEMG_Hyper |
|---|---|---|---|---|---|
| COAD | 11,815 | 10,426 | 4417 | 4886 | 2095 |
| KIRC | 14,087 | 9177 | 2655 | 5325 | 1578 |
| BRCA | 14,667 | 5510 | 4547 | 3228 | 2747 |
| LUSC | 12,147 | 8442 | 4733 | 4040 | 2412 |
| Average-Bioscore (GO-BP) | Average-Bioscore (KEGG) | Average- BHI | # of Cluster | |
|---|---|---|---|---|
| BRCA_hyper | ||||
| Fast Greedy | 0.500 | 0.596 | 0.077 | 27 |
| Infomap | 0.229 | 0.144 | 0.055 | 257 |
| Louvin | 0.400 | 0.422 | 0.069 | 30 |
| COAD_hyper | ||||
| Fast Greedy | 0.289 | 0.427 | 0.069 | 20 |
| Infomap | 0.178 | 0.128 | 0.046 | 227 |
| Louvin | 0.358 | 0.328 | 0.065 | 27 |
| KIRC_hyper | ||||
| Fast Greedy | 0.449 | 0.126 | 0.067 | 15 |
| Infomap | 0.164 | 0.007 | 0.044 | 144 |
| Louvin | 0.342 | 0.05 | 0.056 | 20 |
| LUSC_hyper | ||||
| Fast Greedy | 0.409 | 0.446 | 0.072 | 24 |
| Infomap | 0.135 | 0.032 | 0.049 | 213 |
| Louvin | 0.304 | 0.217 | 0.065 | 31 |
| Average-Bioscore (GO-BP) | Average-Bioscore (KEGG) | Average- BHI | # of Cluster | |
|---|---|---|---|---|
| BRCA_hypo | ||||
| Fast Greedy | 0.427 | 0.539 | 0.081 | 21 |
| Infomap | 0.117 | 0.094 | 0.042 | 274 |
| Louvin | 0.484 | 0.465 | 0.071 | 30 |
| COAD_hypo | ||||
| Fast Greedy | 0.516 | 0.453 | 0.082 | 19 |
| Infomap | 0.132 | 0.064 | 0.042 | 434 |
| Louvin | 0.467 | 0.374 | 0.08 | 35 |
| KIRC_hypo | ||||
| Fast Greedy | 0.525 | 0.499 | 0.083 | 18 |
| Infomap | 0.176 | 0.112 | 0.04 | 377 |
| Louvin | 0.387 | 0.472 | 0.071 | 32 |
| LUSC_hypo | ||||
| Fast Greedy | 0.274 | 0.517 | 0.08 | 25 |
| Infomap | 0.08 | 0.026 | 0.043 | 393 |
| Louvin | 0.525 | 0.351 | 0.074 | 37 |
| Average-Bioscore (GO-BP) | Average-Bioscore (KEGG) | Average- BHI | # of Cluster | |
|---|---|---|---|---|
| BRCA_hyper | ||||
| Fast Greedy | 0.515 | 0.371 | 0.074 | 25 |
| Infomap | 0.187 | 0.096 | 0.066 | 246 |
| Louvin | 0.253 | 0.476 | 0.044 | 32 |
| COAD_hyper | ||||
| Fast Greedy | 0.512 | 0.105 | 0.062 | 17 |
| Infomap | 0.246 | 0.011 | 0.064 | 194 |
| Louvin | 0.359 | 0.057 | 0.050 | 27 |
| KIRC_hyper | ||||
| Fast Greedy | 0.346 | 0.186 | 0.077 | 14 |
| Infomap | 0.191 | 0.106 | 0.071 | 173 |
| Louvin | 0.361 | 0.383 | 0.048 | 22 |
| LUSC_hyper | ||||
| Fast Greedy | 0.460 | 0.349 | 0.074 | 23 |
| Infomap | 0.147 | 0.081 | 0.075 | 220 |
| Louvin | 0.543 | 0.379 | 0.050 | 30 |
| Average-Bioscore (GO-BP) | Average-Bioscore (KEGG) | Average- BHI | # of Cluster | |
|---|---|---|---|---|
| BRCA_hypo | ||||
| Fast Greedy | 0.526 | 0.294 | 0.076 | 23 |
| Infomap | 0.045 | 0.025 | 0.051 | 295 |
| Louvin | 0.299 | 0.243 | 0.077 | 31 |
| COAD_hypo | ||||
| Fast Greedy | 0.305 | 0.567 | 0.089 | 20 |
| Infomap | 0.193 | 0.134 | 0.083 | 424 |
| Louvin | 0.487 | 0.545 | 0.056 | 29 |
| KIRC_hypo | ||||
| Fast Greedy | 0.459 | 0.454 | 0.080 | 21 |
| Infomap | 0.170 | 0.056 | 0.039 | 525 |
| Louvin | 0.415 | 0.199 | 0.074 | 32 |
| LUSC_hypo | ||||
| Fast Greedy | 0.322 | 0.229 | 0.076 | 25 |
| Infomap | 0.087 | 0.036 | 0.070 | 336 |
| Louvin | 0.415 | 0.287 | 0.055 | 33 |
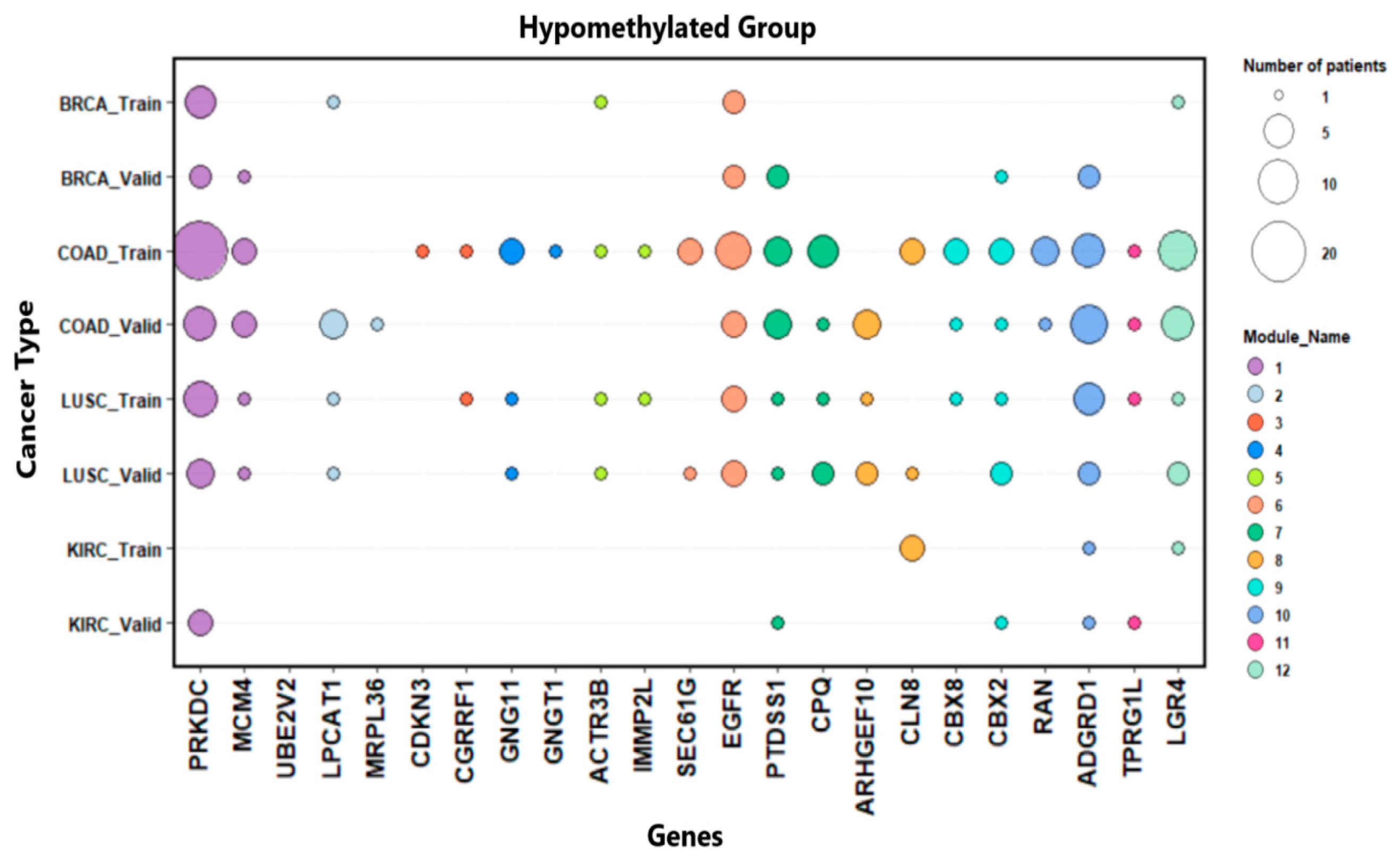
| Genes Name | Methylation Group |
|---|---|
| PRKDC, MCM4, UBE2V2 | Hypo-methylated |
| LPCAT1, mrpl36 | Hypo-methylated |
| CDKN3, CGRRF1 | Hypo-methylated |
| GNG11, GNGT1 | Hypo-methylated |
| ACTR3B, IMMP2L | Hypo-methylated |
| SEC61G, EGFR | Hypo-methylated |
| PTDSS1, CPQ | Hypo-methylated |
| ARHGEF10, CLN8 | Hypo-methylated |
| CBX8, CBX2 | Hypo-methylated |
| RAN, ADGRD1 | Hypo-methylated |
| TPRG1L, PRDM16-DT | Hypo-methylated |
| LGR4, BDNF-AS | Hypo-methylated |
| SLC9A3, PP7080 | Hyper-methylated |
| ENPP5, CYP39A1 | Hyper-methylated |
| RAD54L, EFCAB14 | Hyper-methylated |
| BRIP1, TBX2-AS1 | Hyper-methylated |
| Cancer Type | Average of Low-Level Scores | Average of High-Level Scores |
|---|---|---|
| Brca_hypo | 0.277 | 0.419 |
| Lusc_hypo | 0.282 | 0.41 |
| Coad_hypo | 0.279 | 0.437 |
| Kırc_hypo | 0.276 | 0.381 |
| Brca_hyper | 0.314 | 0.457 |
| Lusc_hyper | 0.285 | 0.434 |
| Coad_hyper | 0.324 | 0.49 |
| Kırc_hyper | 0.364 | 0.489 |
| Cancer Type | Gene Name | Prognostic Score Level | Hazard Rate | p-Value | Number of Patients at Score Level | Number of Deaths |
|---|---|---|---|---|---|---|
| Brca_hypo | GNG11 | Low | 7.7055 | 0.000189 | 11 | 4 |
| CBX2 | High | 2.0370 | 0.0138 | 188 | 27 | |
| Coad_hypo | CDKN3 | High | 2.577 | 0.0262 | 64 | 15 |
| ARHGEF10 | High | 2.855 | 0.0128 | 56 | 14 | |
| GNG11 | High | 2.2279 | 0.0563 | 45 | 12 | |
| CLN8 | High | 3.037 | 0.00823 | 53 | 14 | |
| Kırc_hypo | CBX2 | High | 2.8296 | 0.02 | 19 | 7 |
| Lusc_hypo | SEC61G | High | 1.6608 | 0.0541 | 239 | 99 |
| PTDSS1 | High | 2.6287 | 0.0217 | 273 | 111 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demir Karaman, E.; Işık, Z. Multi-Omics Data Analysis Identifies Prognostic Biomarkers across Cancers. Med. Sci. 2023, 11, 44. https://doi.org/10.3390/medsci11030044
Demir Karaman E, Işık Z. Multi-Omics Data Analysis Identifies Prognostic Biomarkers across Cancers. Medical Sciences. 2023; 11(3):44. https://doi.org/10.3390/medsci11030044
Chicago/Turabian StyleDemir Karaman, Ezgi, and Zerrin Işık. 2023. "Multi-Omics Data Analysis Identifies Prognostic Biomarkers across Cancers" Medical Sciences 11, no. 3: 44. https://doi.org/10.3390/medsci11030044
APA StyleDemir Karaman, E., & Işık, Z. (2023). Multi-Omics Data Analysis Identifies Prognostic Biomarkers across Cancers. Medical Sciences, 11(3), 44. https://doi.org/10.3390/medsci11030044