
Waldenström’s Macroglobulinemia and Cryoglobulinemic Glomerulonephritis: An Unusual Case of Monoclonal Gammopathy of Renal Significance
 , , ,
, , ,
Abstract
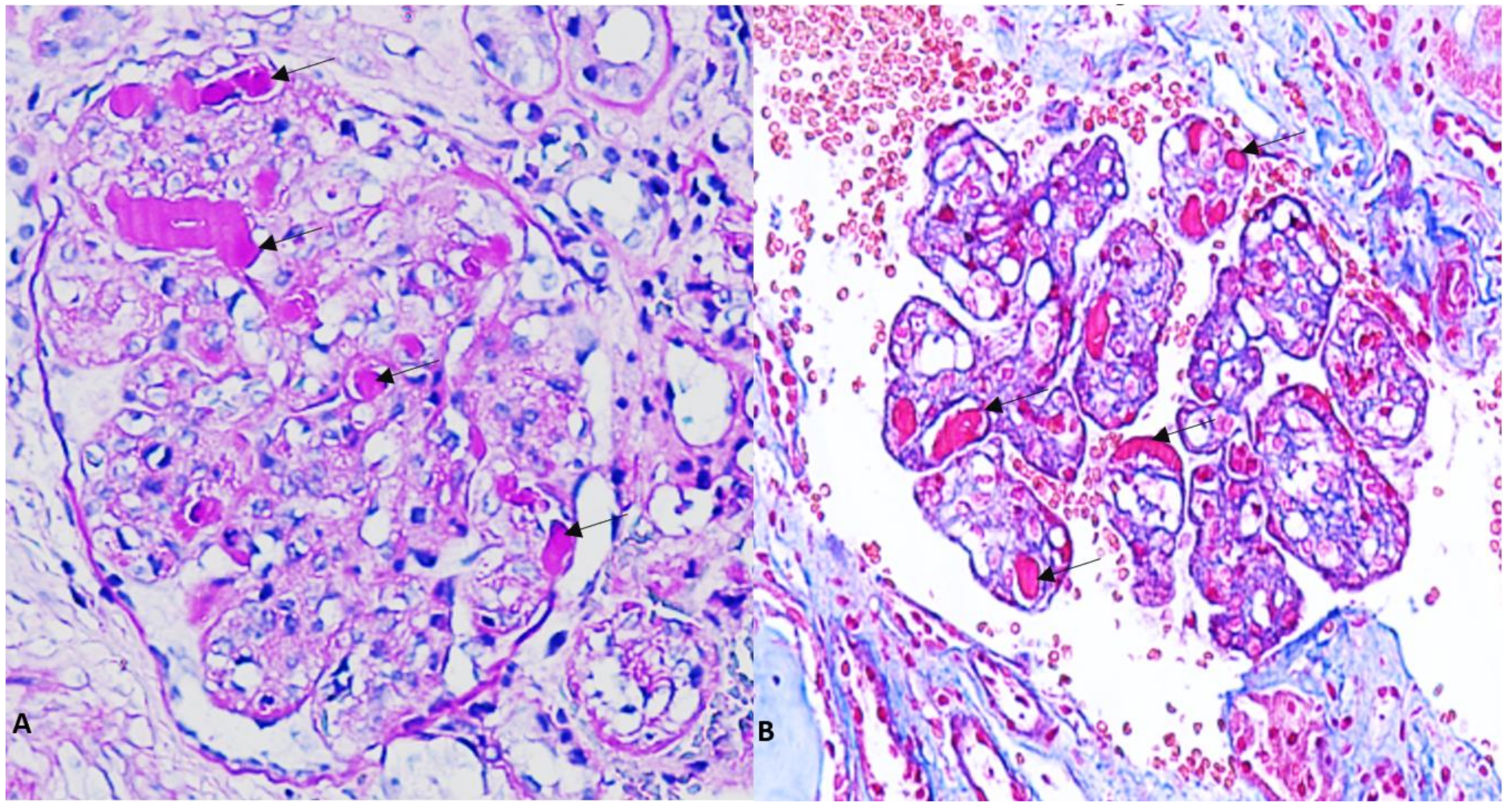
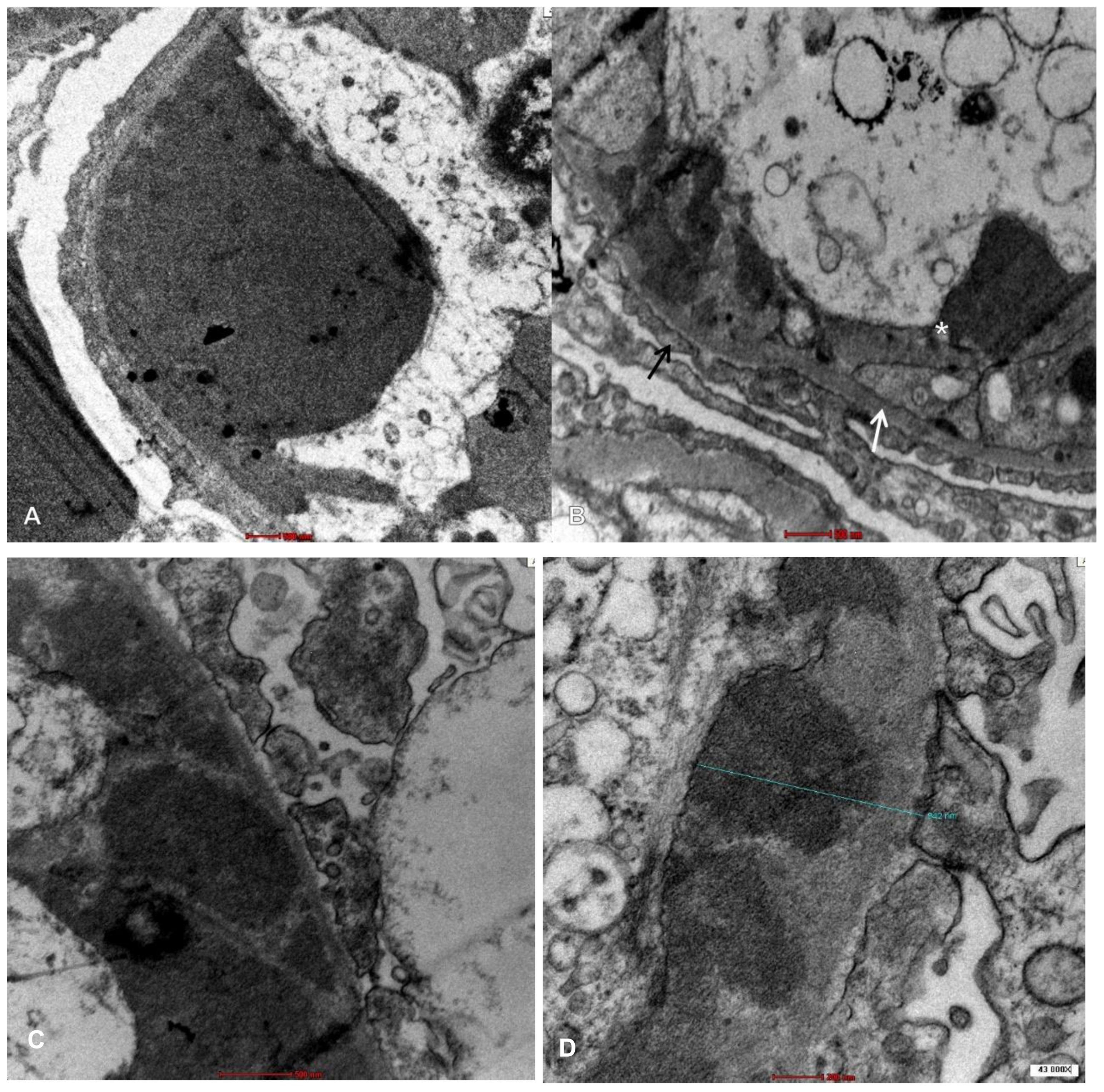
:1. Introduction
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaplan, A.A. Therapeutic apheresis for the renal complications of multiple myeloma and the dysglobulinemias. Ther. Apher. 2001, 5, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Cacoub, P.; Comarmond, C.; Domont, F.; Savey, L.; Saadoun, D. Cryoglobulinemia vasculitis. Am. J. Med. 2015, 128, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Brouet, J.-C.; Clauvel, J.-P.; Danon, F.; Klein, M.; Seligmann, M. Biologic and clinical significance of cryoglobulins. a report of 86 cases. Am. J. Med. 1974, 57, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Ravi, G.; Kapoor, P. Current approach to Waldenström Macroglobulinemia. Cancer Treat. Res. Commun. 2022, 31, 100527. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Kastritis, E. How I treat Waldenstrom macroglobulinemia. Blood 2019, 134, 2022–2035. [Google Scholar] [CrossRef] [PubMed]
- Uppal, N.N.; Monga, D.; Vernace, M.A.; Mehtabdin, K.; Shah, H.H.; Bijol, V.; Jhaveri, K.D. Kidney diseases associated with Waldenström macroglobulinemia. Nephrol. Dial. Transplant. 2019, 34, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Leung, N.; Bridoux, F.; Hutchison, C.A.; Nasr, S.H.; Cockwell, P.; Fermand, J.-P.; Dispenzieri, A.; Song, K.W.; Kyle, R.A. Monoclonal gammopathy of renal significance: When MGUS is no longer undetermined or insignificant. Blood 2012, 120, 4292–4295. [Google Scholar] [CrossRef] [PubMed]
- Roccatello, D.; Saadoun, D.; Ramos-Casals, M.; Tzioufas, A.G.; Fervenza, F.C.; Cacoub, P.; Zignego, A.L.; Ferri, C. Cryoglobulinaemia. Nat. Rev. Dis. Prim. 2018, 4, 11. [Google Scholar] [CrossRef]
- Higgins, L.; Nasr, S.H.; Said, S.M.; Kapoor, P.; Dingli, D.; King, R.L.; Rajkumar, S.V.; Kyle, R.A.; Kourelis, T.; Gertz, M.A.; et al. Kidney involvement of patients with Waldenström macroglobulinemia and other IgM-producing B cell lymphoproliferative disorders. Clin. J. Am. Soc. Nephrol. 2018, 13, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Costenbader, K.; Daikh, D.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.T.; Kamen, D.L.; et al. 2019 European league against rheumatism/American college of rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019, 71, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-P.; Cheng, H.; Rui, H.-L.; Dong, H.-R. Cryoglobulinemic vasculitis and glomerulonephritis: Concerns in clinical practice. Chin. Med. J. 2019, 132, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Chauvet, S.; Bridoux, F.; Ecotière, L.; Javaugue, V.; Sirac, C.; Arnulf, B.; Thierry, A.; Quellard, N.; Milin, S.; Bender, S.; et al. Kidney diseases associated with monoclonal immunoglobulin M–secreting B-cell lymphoproliferative disorders: A case series of 35 patients. Am. J. Kidney Dis. 2015, 66, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, J.; Vos, J.M.I.; Pluimers, T.E.; Japzon, N.; Patel, A.; Salter, S.; Kwakernaak, A.J.; Gupta, R.; Rismani, A.; Kyriakou, C.; et al. Clinical and clonal characteristics of monoclonal immunoglobulin M-associated type I cryoglobulinaemia. Br. J. Haematol. 2023, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.I.; Kouroukis, T.; White, D.; Voralia, M.; Stadtmauer, E.; Wright, J.; Powers, J.; Eisenhauer, E. Bortezomib is active in Waldenstrom’s Macroglobulinemia (WM)—Results of a National Cancer Institute of Canada (NCIC) phase II study in previously untreated or treated WM. J. Clin. Oncol. 2006, 24 (Suppl. 18), 7543. [Google Scholar] [CrossRef]
- Buske, C.; Castillo, J.J.; Abeykoon, J.P.; Advani, R.; Arulogun, S.O.; Branagan, A.R.; Cao, X.; D’Sa, S.; Hou, J.; Kapoor, P.; et al. Report of consensus panel 1 from the 11th International Workshop on Waldenstrom’s Macroglobulinemia on management of symptomatic, treatment-naïve patients. Semin. Hematol. 2023, 60, 73–79. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Normal Range—Units | ||
|---|---|---|
| White blood cells | 8100 | 4–10 × 103 μL |
| Hemoglobin | 9.7 | 12–16 g/dL |
| Platelets | 649 × 103 | 103/μL |
| Reticulocytes | 0.9% | 0.5–2% |
| Lactate dehydrogenase | 126 | 135–214 IU/L |
| Coombs test | Negative | NA |
| Total bilirubin | 0.2 | 0.1–1 mg/dL |
| Total proteins | 4.1 | 6.4–8.7 g/dL |
| Serum albumin (Alb) | 2 | 3–5.5 g/dL |
| AST | 34 | 5–32 IU/L |
| ALT | 7.2 | 5–33 IU/L |
| Urea | 48 | 17–60 mg/dL |
| Creatinine | 0.6 | 0.6–1.2 mg/dL |
| Na+ | 129 | 135–145 mmol/L |
| K+ | 4.8 | 3.5–5.5 mmol/L |
| Cl− | 94 | 95–110 mmol/L |
| C-Reactive protein | Negative | 0.1–0.5 mg/dL |
| Total cholesterol | 324 | mg/dL |
| HBsAg (hepatitis B surface antigen) | Negative | NA |
| Anti-HCV antibodies | Negative | NA |
| HIV | Negative | NA |
| C3 | 69 | 90–180 mg/dL |
| C4 | 2 | 10–40 mg/dL |
| Rheumatoid factor | 9 | <15 IU/mL |
| Beta 2 microglobulin | 2 | <0–20 mg/dL |
| ANA, anti-dsDNA, ANCA | Negative | NA |
| Serum cryoglobulin | Positive | NA |
| Anti-PLA2R antibody (ELISA) | Negative | NA |
| Urine red blood cells | 5–10 | /HPF |
| 24 h urine total protein excretion | 12690.4 | <0.15 g/24-h |
| IgG | 460 | 800–1600 mg/dL |
| IgA | 120 | 70–400 mg/dL |
| IgM | 775 | 90–180 mg/dL |
| UPEP/UIFE | IgM kappa | NA |
| SPEP M-protein concentration | 6% | NA |
| SIFE | IgM kappa | NA |
| FLCκ | 330.16 | 4.90–13.70 mg/L |
| FLCλ | 15.81 | 7.60–19.50 mg/L |
| FLCκ/λ | 20.88 | 0.27–1.67 |
| Urine culture | Negative | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De La Flor, J.C.; Sulca, J.d.M.; Rodríguez, P.; Villa, D.; Sandoval, E.; Zamora, R.; Monroy-Condori, M.; Lipa, R.; Perez, H.; Cieza, M. Waldenström’s Macroglobulinemia and Cryoglobulinemic Glomerulonephritis: An Unusual Case of Monoclonal Gammopathy of Renal Significance. Med. Sci. 2023, 11, 77. https://doi.org/10.3390/medsci11040077
De La Flor JC, Sulca JdM, Rodríguez P, Villa D, Sandoval E, Zamora R, Monroy-Condori M, Lipa R, Perez H, Cieza M. Waldenström’s Macroglobulinemia and Cryoglobulinemic Glomerulonephritis: An Unusual Case of Monoclonal Gammopathy of Renal Significance. Medical Sciences. 2023; 11(4):77. https://doi.org/10.3390/medsci11040077
Chicago/Turabian StyleDe La Flor, José C., Jesús de María Sulca, Pablo Rodríguez, Daniel Villa, Edna Sandoval, Rocío Zamora, Maribel Monroy-Condori, Roxana Lipa, Henry Perez, and Michael Cieza. 2023. "Waldenström’s Macroglobulinemia and Cryoglobulinemic Glomerulonephritis: An Unusual Case of Monoclonal Gammopathy of Renal Significance" Medical Sciences 11, no. 4: 77. https://doi.org/10.3390/medsci11040077