
Disparities in Brain Cancer in the United States: A Literature Review of Gliomas

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Gliomas, Causes, Symptoms, and Classification
1.2. Diagnosis, Treatment, and Prognosis
2. Biology of Gliomas
2.1. Physiology
2.2. Morphology
2.3. Genetics
2.4. Genes
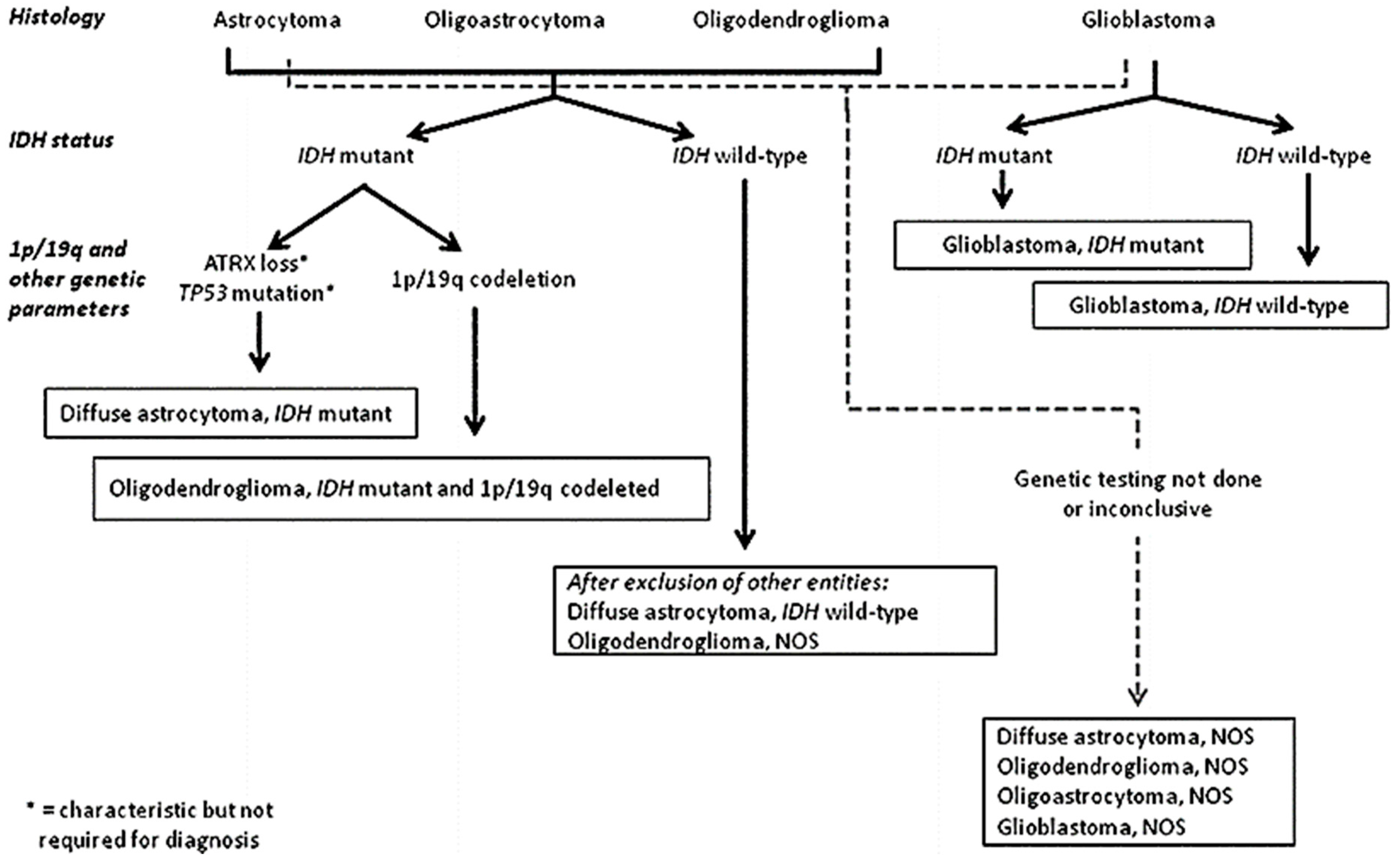
3. Glioma Classification
3.1. Diffuse Astrocytoma and Oligodendroglioma
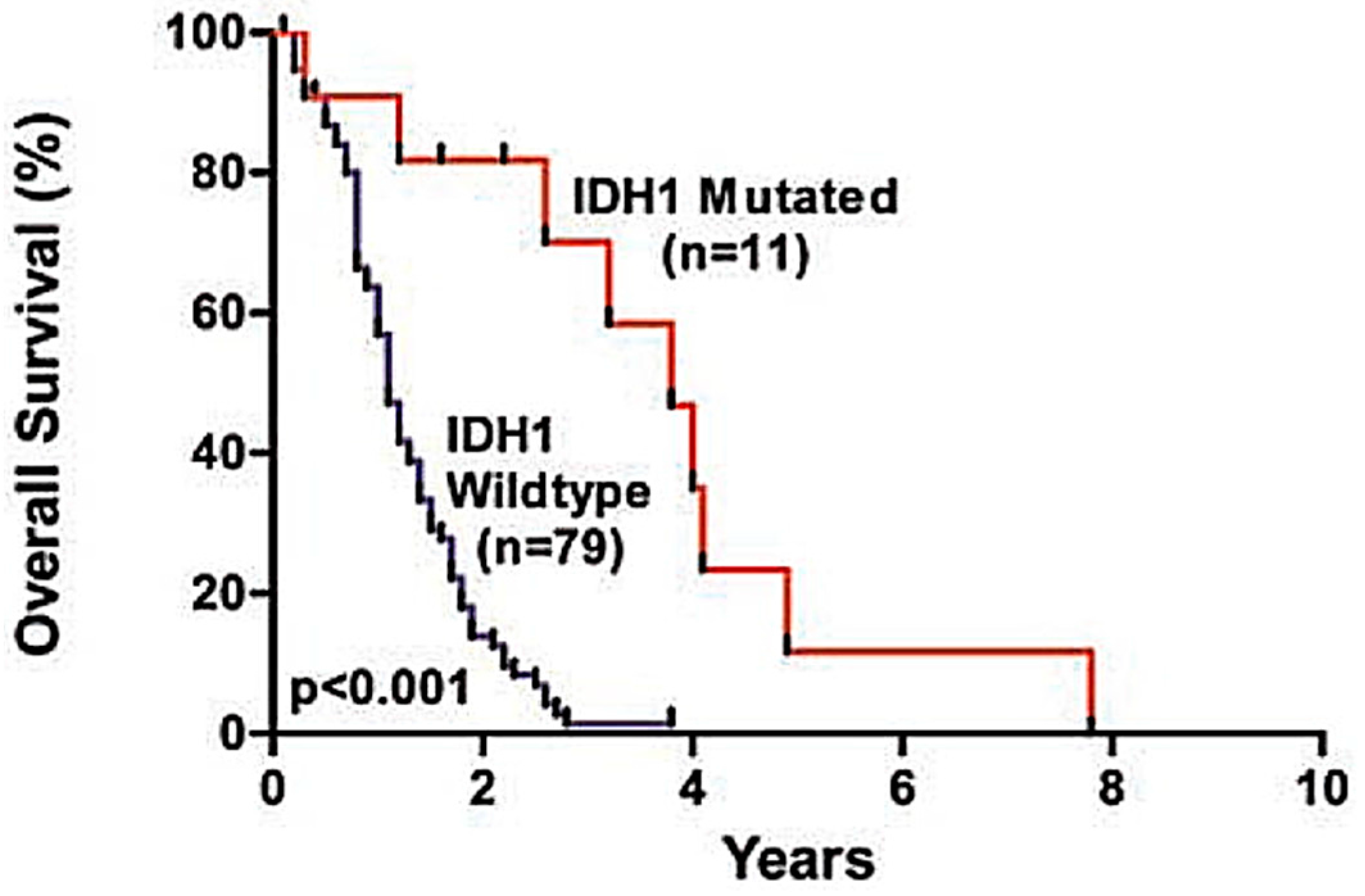
3.1.1. IDH-Mutant Tumours
3.1.2. Nitric Oxidase Synthase Mutation
3.1.3. H3-K27M Mutation and Diffuse Midline Gliomas
3.1.4. 1p/19q Co-Deletion
3.2. Other Astrocytic Tumours
3.3. Ependymal Tumours
3.4. RELA
3.5. Other Gliomas
3.6. Neuronal and Mixed Neuronal Glial Tumours
4. Trends in Gliomas in the United States
4.1. Racial Trends in the United States
4.2. Age and Sex
4.3. Social Determinants
5. Conclusions and Future Work
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ostrom, Q.T.; Gitlleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17 (Suppl. S4), iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Maher, E. Malignant glioma: Genetics and biology of a grave matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef] [PubMed]
- Grier, J.; Batchelor, T. Low-Grade Gliomas in Adults. Oncologist 2006, 11, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Ducray, F.; Idbaih, A.; Wang, X.; Cheneau, C.; Labussiere, M.; Sanson, M. Predictive and prognostic factors for gliomas. Expert Rev. Anticancer Ther. 2011, 11, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Stein, R. Malignant Gliomas Affect About 10,000 Americans Annually. The Washington Post, 20 May 2008. [Google Scholar]
- Komori, T. The 2016 WHO Classification of Tumors of the Central Nervous System: The Major Points of Revision. Neurol. Med. Chir. 2017. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.J.; Burke, A.E.; Glass, R.M. Gliomas. JAMA 2010, 303, 1000. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohqaki, H.; Wiestler, O.T.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumor of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Classification of Tumors of the Central Nervous System, revised 4th ed.; Louis, D.N., Ohgaki, H., Wiestler, O.D., Cavenee, W.K., Eds.; International Agency for Research on Cancer IARC: Lyon, France, 2016. [Google Scholar]
- Blausen Gallery 2014. Wikiversity Journal of Medicine. Available online: https://upload.wikimedia.org/wikipedia/commons/a/a6/Blausen_0870_TypesofNeuroglia.png (accessed on 16 July 2017). [CrossRef]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Wu, Q.; Yan, K.; MacSwords, J.M.; Chandler-Militello, D.; Misuracag, K.L.; Lathia, J.D.; Forrester, M.T.; Lee, J.; Stamler, J.S.; et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell 2011, 146, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol. 2008, 7, 1152–1160. [Google Scholar] [CrossRef]
- Mangani, D.; Weller, M.; Roth, P. The network of immunosuppressive pathways in glioblastoma. Biochem. Pharm. 2017, 130, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zülch, K.J. Histological typing of tumors of the central nervous system; World Health Organization: Geneva, Switzerland, 1979. [Google Scholar]
- Burger, P. Revising the World Health Organization (WHO) Blue Book: Histological Typing of Tumors of the Central Nervous System. J. Neurooncol. 1995, 24, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumors of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Abbas, A.; Aster, J. Robbins Basic Pathology, 9th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2013. [Google Scholar]
- Van den Bent, M.J. Interobserver Variation of The Histopathological Diagnosis in Clinical Trials on Glioma: A Clinician’s Perspective. Acta Neuropathol. 2010, 120, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 Mutations in Gliomas. Cur. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Verhaak, R.J.; Aldape, K.D.; Yung, W.K.; Salama, S.R.; Cooper, L.A.; Rheinbay, E.; Miller, C.R.; Vitucci, M.; Morozova, O.; et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2005, 372, 2481–2498. [Google Scholar]
- Appin, C.; Brat, D.J. Molecular Genetics of Gliomas. Cancer J. 2014, 20, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Sahm, F.; Reuss, D.; Koelsche, C.; Capper, D.; Schittenhelm, J.; Heim, S.; Jones, D.T.; Pfister, S.M.; Herold-Mende, C.; Wick, W.; et al. Farewell to Oligoastrocytoma: In Situ Molecular Genetics Favor Classification as Either Oligodendroglioma or Astrocytoma. Acta Neuropathol. 2014, 128, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Horbinski, C.; Kelly, L.; Nikiforov, Y.E.; Durso, M.B.; Nikiforova, M.N. Detection of IDH1 and IDH2 Mutations by Fluorescence Melting Curve Analysis as a Diagnostic Tool for Brain Biopsies. J. Mol. Diagn. 2010, 12, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Appin, C.L.; Daniel, J.B. Molecular Pathways in Gliomagenesis and Their Relevance to Neuropathologic Diagnosis. Adv. Anat. Pathol. 2015, 22, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Verkhratsky, A. Neuroglia—Living Nerve Glue. Fortschritte Neurologie Psychiatrie 2011, 79, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Paleologos, N. Astrocytoma. In About Brain Tumors: A Primer for Patients and Caregivers, 1st ed.; American Brain Tumor Assocation, Ed.; American Brain Tumor Association: Chicago, IL, USA, 2016. [Google Scholar]
- Radiopedia; Jones, J.; Gaillard, F.; et al. Glioblastoma. Available online: https://radiopaedia.org/articles/glioblastoma (accessed on 27 March 2017).
- Wikipedia. Glioblastoma Multiforme. Available online: https://commons.wikimedia.org/wiki/File:Glioblastoma_(1).jpg (accessed on 18 July 2017).
- Wikipedia. Glioblastoma GFAP. Available online: https://commons.wikimedia.org/wiki/File:Glioblastoma_GFAP.jpg (accessed on 27 March 2017).
- Bredt, D.; Snyder, S. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Hara, A. Cyclooxygenase-2 and inducible nitric oxide synthase expression in human astrocytic gliomas: Correlation with angiogenesis and prognostic significance. Acta Neuropathol. 2004, 108, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Schartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quanq, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.; Wood, M.; Tihan, T.; Bollen, A.; Gupta, N.; Phillips, J.; Perry, A. Diffuse midline gliomas with Histone H3-K27M Mutation: A series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol. 2016, 26, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Ryzhova, M.; Hovestadt, V.; Bender, S.; Sturm, D.; Capper, D.; Meyer, J.; Schrimpf, D.; Kool, M.; Northcott, P.A.; et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015, 129, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed]
- López, G.; Ann, N.; Bush, O.; Berger, M.S.; Perry, A.; Solomon, D.A. Diffuse Non-midline Glioma with H3F3A K27M Mutation: A Prognostic and Treatment Dilemma. Acta Neuropathol. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.; Sethares, C. The Fine Structure of the Aging Brain, 1st ed.; Boston University Press: Boston, MA, USA; Available online: https://www.bu.edu/agingbrain/download-page/ (accessed on 27 March 2017).
- Paleologos, N. Oligodendroglioma and Oligoastrocytoma. In About Brain Tumors: A Primer for Patients and Caregivers, 1st ed.; American Brain Tumor Assocation, Ed.; American Brain Tumor Association: Chicago, IL, USA, 2016. [Google Scholar]
- McCarthy, B.; Rankin, K.; Aldape, K.; Bondy, M.L.; Brännström, T.; Broholm, H.; Feychting, M.; Il’yasova, D.; Inskip, P.D.; Johansen, C.; et al. Risk factors for oligodendroglial tumors: A pooled international study. Neuro Oncol. 2010, 13, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Van den Bent, M.J.; Smits, M.; Kros, J.M.; Chang, S.M. Diffuse Infiltrating Oligodendroglioma and Astrocytoma. J. Clin. Oncol. 2017, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Scheie, D.; Meling, T.; Cvancarova, M.; Skullerud, K.; Mørk, S.; Lote, K.; Eide, T.J.; Helseth, E.; Beiske, K. Prognostic variables in oligodendroglial tumors: A single-institution study of 95 cases. Neuro Oncol. 2011, 13, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Wikipedia. Oligodendroglioma. Available online: https://commons.wikimedia.org/wiki/File:Oligodendroglioma_001.jpg (accessed on 18 July 2017).
- Wikipedia. Glioma Gross 4. Available online: https://www.wikidoc.org/index.php/File:Glioma_Gross_4.jpg (accessed on 18 July 2017).
- World Health Organization Classification of Tumors. A Public Database of Human Cancers. Available online: http://www.pubcan.org/cancer/18/oligodendroglioma-nos/histopathology (accessed on 27 March 2017).
- Fernandez, C.; Figarella-Branger, D.; Girard, N.; Bouvier-Labit, C.; Gouvernet, J.; Paz Paredes, A.; Lena, G. Pilocytic Astrocytomas in Children: Prognostic Factors—A Retrospective Study of 80 Cases. Neurosurgery 2003, 53, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Pathologyoutlines. CNS Tumor—Subependymoma. Available online: http://www.pathologyoutlines.com/topic/cnstumorsubependymoma.html (accessed on 1 April 2017).
- Koeller, K.K.; Sandberg, G.D. From the Archives of the AFIP. Cerebral Intraventricular neoplasms: radiologic-pathologic correlation. Radiographics 2002, 6, 1473–1505. [Google Scholar] [CrossRef] [PubMed]
- Pathologyoutlines. CNS Tumor—Myxopapillary Ependymoma. Available online: http://www.pathologyoutlines.com/topic/cnstumormyxopapillaryependymoma.html (accessed on 27 March 2017).
- Radiopaedia. Spine Ependymomas. Available online: https://radiopaedia.org/articles/spinal-ependymoma (accessed on 27 March 2017).
- Radiopaedia. Ependymoma. Available online: https://radiopaedia.org/cases/ependymoma-lateral-ventricle (accessed on 27 March 2017).
- University of Cape Town Digital Pathology Teaching Collection. Hemisphere Ependymoma. Available online: http://www.digitalpathology.uct.ac.za/catalogue_detail.php?case_id=2429&discipline_id=6 (accessed on 18 July 2017).
- CERN Foundation. Adult Ependymoma Images. Available online: http://www.cern-foundation.org/education/diagnosis/adult-ependymoma-images (accessed on 27 March 2017).
- Wikipedia. Ependymoma Pseudorosette. Available online: https://commons.wikimedia.org/wiki/File:Ependymoma_pseudorosette.jpg (accessed on 18 July 2017).
- Jakacki, R. Ependymoma. In About Brain Tumors: A Primer for Patients and Caregivers, 10th ed.; American Brain Tumor Association: Chicago, IL, USA, 2016. [Google Scholar]
- Armstrong, T.; Vera-Bolanos, E.; Bekele, B.; Aldape, K.; Gilbert, M. Adult ependymal tumors: Prognosis and the M. D. Anderson Cancer Center experience. Neuro Oncol. 2010, 12, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Horn, B.; Heideman, R.; Geyer, R.; Pollack, I.; Packer, R.; Goldwein, J.; Tomita, T.; Schomberg, P.; Ater, J.; et al. A Multi-Institutional Retrospective Study of Intracranial Ependymoma in Children identification of risk factors. J. Pediatr. Hematol. Oncol. 1999, 21, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.; Mohankumar, K.; Punchihewa, C.; Weinlich, R.; Dalton, J.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.M.; Thiruvenkatam, R.; et al. C11orf95–RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 2014, 506, 451–455. [Google Scholar] [CrossRef] [PubMed]
- McCracken, J.A.; Gonzales, M.F.; Phal, P.M.; Drummond, K.J. Angiocentric glioma transformed into anaplastic ependymoma: Review of the evidence for malignant potential. J. Clin. Neurosci. 2016, 34, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Alexandru, D.; Haghighi, B.; Muhonen, M.G. The treatment of angiocentric glioma: Case report and literature review. Perm J. 2013, 17, e100–e102. [Google Scholar] [CrossRef] [PubMed]
- Samkari, A.; Hmoud, M.; Al-Mehdar, A.; Abdullah, S. Well-differentiated and anaplastic astroblastoma in the same patient: A case report and review of the literature. Clin. Neuropathol. 2015, 34, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Ampie, L.; Choy, W.; Lamano, J.B.; Kesavabhotla, K.; Mao, Q.; Parsa, A.T.; Bloch, O. Prognostic factors for recurrence and complications in the surgical management of primary chordoid gliomas: A systematic review of literature. Clin. Neurol. Neursurg. 2015, 138, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Deorah, S.; Lynch, C.; Sibenaller, Z.; Ryken, T. Trends in brain cancer incidence and survival in the United States: Surveillance, Epidemiology, and End Results Program, 1973 to 2001. Neurosurg. Focus 2006, 20, E1. [Google Scholar] [CrossRef] [PubMed]
- Dubrow, R.; Darefsky, A. Demographic variation in incidence of adult glioma by subtype, United States, 1992–2007. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed]











© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Persaud-Sharma, D.; Burns, J.; Trangle, J.; Moulik, S. Disparities in Brain Cancer in the United States: A Literature Review of Gliomas. Med. Sci. 2017, 5, 16. https://doi.org/10.3390/medsci5030016
Persaud-Sharma D, Burns J, Trangle J, Moulik S. Disparities in Brain Cancer in the United States: A Literature Review of Gliomas. Medical Sciences. 2017; 5(3):16. https://doi.org/10.3390/medsci5030016
Chicago/Turabian StylePersaud-Sharma, Dharam, Joseph Burns, Jeran Trangle, and Sabyasachi Moulik. 2017. "Disparities in Brain Cancer in the United States: A Literature Review of Gliomas" Medical Sciences 5, no. 3: 16. https://doi.org/10.3390/medsci5030016
APA StylePersaud-Sharma, D., Burns, J., Trangle, J., & Moulik, S. (2017). Disparities in Brain Cancer in the United States: A Literature Review of Gliomas. Medical Sciences, 5(3), 16. https://doi.org/10.3390/medsci5030016